Development of a Soft Cell Confiner to Decipher the Impact of Mechanical Stimuli on Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

AC/DC You Shook Me All Night Long Adele Rolling in the Deep Al Green
AC/DC You Shook Me All Night Long Adele Rolling in the Deep Al Green Let's Stay Together Alabama Dixieland Delight Alan Jackson It's Five O'Clock Somewhere Alex Claire Too Close Alice in Chains No Excuses America Lonely People Sister Golden Hair American Authors The Best Day of My Life Avicii Hey Brother Bad Company Feel Like Making Love Can't Get Enough of Your Love Bastille Pompeii Ben Harper Steal My Kisses Bill Withers Ain't No Sunshine Lean on Me Billy Joel You May Be Right Don't Ask Me Why Just the Way You Are Only the Good Die Young Still Rock and Roll to Me Captain Jack Blake Shelton Boys 'Round Here God Gave Me You Bob Dylan Tangled Up in Blue The Man in Me To Make You Feel My Love You Belong to Me Knocking on Heaven's Door Don't Think Twice Bob Marley and the Wailers One Love Three Little Birds Bob Seger Old Time Rock & Roll Night Moves Turn the Page Bobby Darin Beyond the Sea Bon Jovi Dead or Alive Living on a Prayer You Give Love a Bad Name Brad Paisley She's Everything Bruce Springsteen Glory Days Bruno Mars Locked Out of Heaven Marry You Treasure Bryan Adams Summer of '69 Cat Stevens Wild World If You Want to Sing Out CCR Bad Moon Rising Down on the Corner Have You Ever Seen the Rain Looking Out My Backdoor Midnight Special Cee Lo Green Forget You Charlie Pride Kiss an Angel Good Morning Cheap Trick I Want You to Want Me Christina Perri A Thousand Years Counting Crows Mr. -
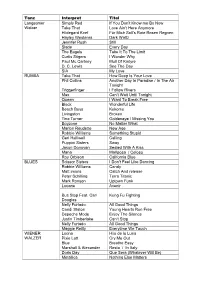
Tanz Interpret Titel Langsamer Walzer Simply Red If You Don't Know Me
Tanz Interpret Titel Langsamer Simply Red If You Don’t Know me By Now Walzer Take That Love Ain’t Here Anymore Hildegard Knef Für Mich Soll’s Rote Rosen Regnen Hayley Westenra Dark Waltz Jennifer Rush Still Slade Every Day The Eagels Take It To The Limit Curtis Stigers I Wonder Why Paul Mc Cartney Mull Of Kintyre D. C. Lewis See The Day SIA My Love RUMBA Take That How Deep Is Your Love Phil Collins Another Day In Paradise / In The Air Tonight Triggerfinger I Follow Rivers Max Can’t Wait Until Tonight Queen I Want To Break Free Black Wonderful Life Beach Boys Kokomo Livingston Broken Tina Turner Goldeneye / Missing You Boyzone No Matter What Marlon Roudette New Age Robbie Williams Something Stupid Geri Halliwell Calling Puppini Sisters Sway Jason Donovan Sealed With A Kiss Mana Mariposa / Celoso Roy Orbison California Blue BLUES Scissor Sisters I Don’t Feel Like Dancing Robbie Williams Candy Matt imons Catch And release Peter Schilling Terra Titanic Mark Ronson Uptown Funk Louane Avenir Bus Stop Feat. Carl Kung Fu Fighting Douglas Nelly Furtado All Good Things Candi Staton Young Hearts Run Free Depeche Mode Enjoy The Silence Justin Timberlake Can’t Stop Nelly Furtado All Good Things Maggie Reilly Everytime We Touch WIENER Loona Hijo de la Luna WALZER Pixie Lott Cry Me Out Blue Breathe Easy Marshall & Alexander Resta / In Italy Doris Day Que Sera (Whatever Will Be) Metallica Nothing Else Matters The Corrs Runaway Kelly Clarkson Breakaway Nelly Furtado In Gods Hands Kylie Minogue & Nick Where The Wild Roses Grow Caven CHA CHA CHA Cher Shoop Shoop Song Caro Emerald A Night Like This Peter Fox Haus Am See Debelah Morgan Dance With Me Mana El Rey Tiburon Hot Banditoz Veo Veo Eruption One Way Ticket DNCE Cake By The Ocean Robin Thicke Blurred Lines Carlos Santana Smooth The Pussycat Dolls Perhaps Perhaps Perhaps Carlos Santana Corazon Espinado / Smooth Marc Anthony I Need To Know Jennifer Lopez Let’s Get Loud Gotye Feat. -
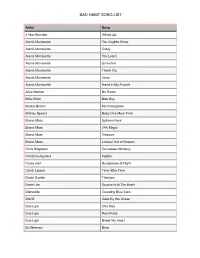
Bad Habit Song List
BAD HABIT SONG LIST Artist Song 4 Non Blondes Whats Up Alanis Morissette You Oughta Know Alanis Morissette Crazy Alanis Morissette You Learn Alanis Morissette Uninvited Alanis Morissette Thank You Alanis Morissette Ironic Alanis Morissette Hand In My Pocket Alice Merton No Roots Billie Eilish Bad Guy Bobby Brown My Prerogative Britney Spears Baby One More Time Bruno Mars Uptown Funk Bruno Mars 24K Magic Bruno Mars Treasure Bruno Mars Locked Out of Heaven Chris Stapleton Tennessee Whiskey Christina Aguilera Fighter Corey Hart Sunglasses at Night Cyndi Lauper Time After Time David Guetta Titanium Deee-Lite Groove Is In The Heart Dishwalla Counting Blue Cars DNCE Cake By the Ocean Dua Lipa One Kiss Dua Lipa New Rules Dua Lipa Break My Heart Ed Sheeran Blow BAD HABIT SONG LIST Artist Song Elle King Ex’s & Oh’s En Vogue Free Your Mind Eurythmics Sweet Dreams Fall Out Boy Beat It George Michael Faith Guns N’ Roses Sweet Child O’ Mine Hailee Steinfeld Starving Halsey Graveyard Imagine Dragons Whatever It Takes Janet Jackson Rhythm Nation Jessie J Price Tag Jet Are You Gonna Be My Girl Jewel Who Will Save Your Soul Jo Dee Messina Heads Carolina, Tails California Jonas Brothers Sucker Journey Separate Ways Justin Timberlake Can’t Stop The Feeling Justin Timberlake Say Something Katy Perry Teenage Dream Katy Perry Dark Horse Katy Perry I Kissed a Girl Kings Of Leon Sex On Fire Lady Gaga Born This Way Lady Gaga Bad Romance Lady Gaga Just Dance Lady Gaga Poker Face Lady Gaga Yoü and I Lady Gaga Telephone BAD HABIT SONG LIST Artist Song Lady Gaga Shallow Letters to Cleo Here and Now Lizzo Truth Hurts Lorde Royals Madonna Vogue Madonna Into The Groove Madonna Holiday Madonna Border Line Madonna Lucky Star Madonna Ray of Light Meghan Trainor All About That Bass Michael Jackson Dirty Diana Michael Jackson Billie Jean Michael Jackson Human Nature Michael Jackson Black Or White Michael Jackson Bad Michael Jackson Wanna Be Startin’ Something Michael Jackson P.Y.T. -

Soft Cell Tainted Love Mp3, Flac, Wma
Soft Cell Tainted Love mp3, flac, wma DOWNLOAD LINKS (Clickable) Genre: Electronic Album: Tainted Love Country: Canada Released: 1981 Style: Synth-pop MP3 version RAR size: 1901 mb FLAC version RAR size: 1643 mb WMA version RAR size: 1552 mb Rating: 4.4 Votes: 514 Other Formats: DXD WAV ASF WMA ADX AU XM Tracklist A Tainted Love 2:41 B Tainted Dub 4:50 Companies, etc. Manufactured By – Polygram Inc. Distributed By – Polygram Distribution Inc. Phonographic Copyright (p) – Some Bizzare Label Credits Engineer – Paul Hardiman Producer – Mike Thorne Written-By – Ed Cobb Notes Cover-Version of Gloria Jones - Tainted Love (New U.S. Remix) (Nouveau Mixage U.S.) A side label: "Original Sound Recording make by Some Bizzare Label" B side label: "Original Sound Recording made by Some Bizzare Label" ℗ 1981 Some Bizzare Label Taken from the Vertigo Album & Cassette Soft Cell - Non-Stop Erotic Cabaret Made in Canada Also available with a Soft Cell - Tainted Love slightly different label design and info. Barcode and Other Identifiers Matrix / Runout (Side A): SOV-2304-A Matrix / Runout (Side B): SOV-2304-B Other versions Category Artist Title (Format) Label Category Country Year BZS 2 Soft Cell Tainted Love (7", Single, Pap) Some Bizzare BZS 2 UK 1981 Tainted Love = Amor Incierto (7", 6059527 Soft Cell Vertigo 6059527 Argentina 1982 Single) 6059 448 Soft Cell Tainted Love (7", Single) Vertigo 6059 448 Austria 1981 6059 448 Soft Cell Tainted Love (7", Single) Vertigo 6059 448 Italy 1981 Tainted Love / Where Did Our Love 6359 081 Soft Cell Mercury 6359 081 Australia 1981 Go (12") Related Music albums to Tainted Love by Soft Cell Soft Cell - Nonstop Ecstatic Dancing Tainted DickMen - Tainted DickMen Soft Cell - Non Stop Ecstatic Dancing Soft Cell - Tainted Love / Memorabilia Soft Cell, Marc Almond - Tainted Love Soft Cell / Marc Almond - Tainted Love '91 Soft Cell - Torch Soft Cell / Marc Almond - Memorabilia - The Singles Soft Cell - Down In The Subway Gloria Jones - Tainted Love Soft Cell - What! Soft Cell - Soft Cell's Non-Stop Exotic Video Show. -

Most Requested Songs of 2009
Top 200 Most Requested Songs Based on nearly 2 million requests made at weddings & parties through the DJ Intelligence music request system in 2009 RANK ARTIST SONG 1 AC/DC You Shook Me All Night Long 2 Journey Don't Stop Believin' 3 Lady Gaga Feat. Colby O'donis Just Dance 4 Bon Jovi Livin' On A Prayer 5 Def Leppard Pour Some Sugar On Me 6 Morrison, Van Brown Eyed Girl 7 Beyonce Single Ladies (Put A Ring On It) 8 Timberlake, Justin Sexyback 9 B-52's Love Shack 10 Lynyrd Skynyrd Sweet Home Alabama 11 ABBA Dancing Queen 12 Diamond, Neil Sweet Caroline (Good Times Never Seemed So Good) 13 Black Eyed Peas Boom Boom Pow 14 Rihanna Don't Stop The Music 15 Jackson, Michael Billie Jean 16 Outkast Hey Ya! 17 Sister Sledge We Are Family 18 Sir Mix-A-Lot Baby Got Back 19 Kool & The Gang Celebration 20 Cupid Cupid Shuffle 21 Clapton, Eric Wonderful Tonight 22 Black Eyed Peas I Gotta Feeling 23 Lady Gaga Poker Face 24 Beatles Twist And Shout 25 James, Etta At Last 26 Black Eyed Peas Let's Get It Started 27 Usher Feat. Ludacris & Lil' Jon Yeah 28 Jackson, Michael Thriller 29 DJ Casper Cha Cha Slide 30 Mraz, Jason I'm Yours 31 Commodores Brick House 32 Brooks, Garth Friends In Low Places 33 Temptations My Girl 34 Foundations Build Me Up Buttercup 35 Vanilla Ice Ice Ice Baby 36 Bee Gees Stayin' Alive 37 Sinatra, Frank The Way You Look Tonight 38 Village People Y.M.C.A. -

The UK Top 200 Most Requested Songs in 2018 This List Is Compiled Based on Over 2 Million Song Requests Made Using the DJ Event Planner Song Request System
The UK Top 200 Most Requested Songs In 2018 This list is compiled based on over 2 million song requests made using the DJ Event Planner song request system. Rank Artist Song Title 1 Killers Mr. Brightside 2 Whitney Houston I Wanna Dance With Somebody (Who Loves Me) 3 Mark Ronson feat. Bruno Mars Uptown Funk 4 Bon Jov i Livin' On A Prayer 5 ABBA Dancing Queen 6 Walk The Moon Shut Up And Dance 7 Bryan Adams Summer Of '69 8 Kings Of Leon Sex On Fire 9 Pharrell Williams Happy 10 Black Eyed Peas I Gotta Feeling 11 Dexys Midnight Runners Come On Eileen 12 Justin Timberlake Can't Stop The Feeling! 13 Luis Fonsi feat. Daddy Yankee Despacito 14 Journey Don't Stop Believin' 15 Queen Don't Stop Me Now 16 Bruno Mars Marry You 17 Neil Diamond Sweet Caroline 18 Oasis Wonderwall 19 Toploader Dancing In The Moonlight 20 Beyonce feat. Jay-Z Crazy In Love 21 Beyonce Single Ladies (Put A Ring On It) 22 Arctic Monkeys I Bet You Look Good On The Dancefloor 23 Rihanna feat. Calv in Harris We Found Love 24 DJ Casper Cha Cha Slide 25 Wham! Wake Me Up Before You Go-go 26 Taylor Swift Shake It Off 27 Ed Sheeran Shape Of You 28 Mark Ronson feat. Amy Winehouse Valerie 29 Van Morrison Brown Eyed Girl 30 Foundations Build Me Up Buttercup 31 Cyndi Lauper Girls Just Want To Have Fun 32 John Trav olta & Oliv ia Newton-John Grease Megamix 33 R. Kelly Ignition (Remix) 34 September 34 Earth, Wind and Fire September 35 B-52's Love Shack 36 Los Del Rio Macarena 37 Guns N' Roses Sweet Child O' Mine 38 Kenny Loggins Footloose 39 Maroon 5 Moves Like Jagger 40 Bill Medley & Jennifer Warnes (I've Had) The Time Of My Life 41 Stev ie Wonder Superstition 42 Creedence Clearwater Rev iv al / Tina Turner Proud Mary 43 Snap! Rhythm Is A Dancer 44 Usher Yeah! 45 Spice Girls Wannabe 46 OutKast Hey Ya! 47 Robin S Show Me Love 48 Michael Jackson Billie Jean 49 House Of Pain Jump Around 50 Beatles Twist And Shout 51 Village People Y.M.C.A. -

CLASSIC SONGS (60S/70S/80S/90S)
MODERN SONGS (2000 – today) ARTIST SONG ARTIST SONG Adele Someone Like You John Mayer Waiting On The World To Change Adele Rolling In The Deep John Mayer Gravity Adele Set Fire To The Rain John Mayer Slow Dancing In A Burning Room John Mayer Stop This Train Alicia Keys If I Ain’t Got You Aloe Blacc I Need A Dollar Justin Timberlake Suit & Tie Amy Winehouse Rehab Justin Timberlake Rock Your Body Amy Winehouse Valerie Justin Timberlake Senorita Angus & Julia Stone Big Jet Plane Kings Of Leon Use Somebody Ben Harper Steal My Kisses Kings Of Leon Sex On Fire Beyonce Love On Top LMFAO Party Rocking Bruno Mars Just The Way You Are LMFAO Sexy And I Know It Bruno Mars Locked Out Of Heaven Maroon 5 This Love Bruno Mars Lazy Song Maroon 5 Moves Like Jagger Carly Rae Jepsen Call Me Maybe Maroon 5 She Will Be Loved Cee Lo Green Forget You Maroon 5 Sunday Morning Daniel Merriweather Change Michael Buble Home David Guetta feat.Sia Titanium Michael Buble Everything David Gray The One I Love Michael Buble How Sweet It Is Ed Sheeran The A-Team Mumford & Sons The Cave Foster The People Pumped Up Kicks Mumford & Sons Little Lion Man Foster The People Call It What You Want Neyo Closer Frank Ocean Thinkin Bout You Neyo So Sick Frank Ocean Sweet Life Norah Jones Come Away With Me fun. We Are Young Norah Jones Don’t Know Why George Michael Amazing Outkast Hey Ya Gnarls Barkley Crazy Outkast Roses Gotye Heart's A Mess Paolo Nutini Jenny Don't Be Hasty Gotye I Feel Better Pete Murray Feeler Gotye In Your Light Pete Murray So Beautiful Gotye Somebody That I Used -

Song Title Artist Votes Dancing Queen Abba 6 Disco Inferno The
Song Title Artist Votes Dancing Queen Abba 6 Disco Inferno The Trammps 5 Get Lucky Daft Punk 4 Rock the Casbah The Clash 4 Night Fever Bee Gees 3 Le Freak Chic 3 Viva La Vida Coldplay 3 Uptown Funk Mark Ronson ft Bruno Mars 3 Tainted Love Soft Cell 3 Don't Leave me this Way The Communards 3 Town Called Malice The Jam 3 Thunderstruck ACDC 2 Valerie Amy Winehouse 2 Lip up Fatty Bad Manners 2 Twist and Shout Beatles 2 Staying Alive Bee Gees 2 Crazy in Love Beyonce 2 Call Me Blondie 2 One Way or Another Blondie 2 Dancing in the Dark Bruce Springsteen 2 Word Up Cameo 2 Walking on Sunshine Catrina and the Waves 2 I Feel For You Chaka Khan 2 Life on Mars David Bowie 2 You Spin Me Round Dead or Alive 2 Boom! Shake the Room DJ Jazzy Jeff & the Fresh Prince 2 Rio Duran Duran 2 Boogie Wonderland Earth Wind and Fire 2 Let's Groove Earth Wind and Fire 2 American Boy Estelle 2 Rockafeller Skank Fatboy Slim 2 The Love I Lost Harold Melvin & The Blue Notes 2 You Sexy Thing Hot Chocolate 2 (Your Love Keeps Lifting Me) HigherJackie and Wilson Higher 2 Price Tag Jessie J 2 I Predict a Riot Kaiser Chiefs 2 Hot and Cold Katy Perry 2 Somewhere Only We Know Keane 2 Sex on Fire Kings of Leon 2 Celebration Kool & the Gang 2 Sexy and I Know It LMFAO 2 Movin On Up M People 2 Baggy Trousers Madness 2 Moves Like Jagger Maroon 5 2 Sugar Maroon 5 2 Aint no Stopping Us Know Mcfadden & Whitehead 2 Billie Jean Michael Jackson 2 Blue Monday New Order 2 You Can't Hurry Love Phil Collins 2 Disco 2000 Pulp 2 Ready to Go Republica 2 Rock DJ Robbie Williams 2 Don't You Forget -

Songs Always Something There to Remind
Flashback Heart Attack - Songs Always Something There to Remind Me - Naked Eyes Anything Anything – Dramarama Any Way You Want It – Journey Bad Luck - Social Distortion Bizarre Love Triangle - New Order Blister in Sun/Add it Up/Gone Daddy Gone (Medley) - Violent Femmes Blitzkrieg Bop –Ramones Blue Monday - New Order Boys Don’t Cry – The Cure Centerfold - J Giles Band Come on Eileen – Dexies Midnight Runner Dancing w Myself - Billy Idol Destination Unknown – Missing Persons Don’t Change - INXS Don’t Stop Belivin – Journey Don’t You Forget About Me - Simple Minds Don’t You Want Me – Human League Dream Police - Cheap Trick Erotic City - Prince Everybody Wants to Rule the World - Tears for Fears Eyes without a Face – Billy Idol Fascination - Human League Fight for Your Right to Party – Beastie Boys Girls Just Wanna Have Fun – Cyndi Lauper Hungry Like the Wolf – Duran Duran I Love Rock n Roll - Joan Jett I Ran - A Flock of Seagulls In Cars - Gary Newman Jenny Jenny (867-5309) - Tommy Tutone Jesses Girl - Rick Springfield Just Can’t Get Enough - Depeche Mode Just Like Heaven - The Cure Just What I Needed - The Cars Land Down Under – Men at Work Let’s Go Crazy – Prince Live and Let Die – Wings Living on a Prayer – Bon Jovi Mexican Radio – Wall of VooDoo Melt With You - Modern English Metro - Berlin Million Miles Away – The Plimsouls Mirror in the Bathroom – English Beat Modern Love - David Bowie Need You Tonight - INXS Only the Lonely – The Motels Personal Jesus - Depeche Mode Pretty In Pink – Psychedelic Furs Promises Promises – Naked Eyes -

Host's Master Artist List for Game 1
Host's Master Artist List for Game 1 You can play the list in the order shown here, or any order you prefer. Tick the boxes as you play the songs. Donny Osmond Byrds Dizzie Rascal Curtis Stigers Curiosity Killed The Cat Shakira Deacon Blue Eruption Gina G Louis Armstrong Black Grape Midnight Oil Cndigans Bluetones Irene Cara U2 Emili Sande Jimi Hendrix Mel & Kim Thin Lizzy Surviver Estelle Joy Division John Legend Bill Haley Space NSYNC Desmond Dekker Specials REM Men At Work Was Not Was Rihanna George Harrison Cher ZZ Top Ce Ce Peniston Billy Ray Cyrus Gorillaz Bow Bow Bow Chubby Checker Morrissey Alexander O'Neal Soft Cell Beastie Boys The Fray OMD Musical Youth Avicii Beatles Copyright QOD Page 1/38 Host's Master Artist List for Game 2 You can play the list in the order shown here, or any order you prefer. Tick the boxes as you play the songs. The Knack Brotherhood of Man Frankie Valli Earth Wind & Fire Pink Brother Beyond Billy J Kramer Gloria Estefan B Witched Basshunter Feeder Contours Another Level Wilson Phillips Barry Manilow Aztec Camera Good Charlotte Elvis Presley Foo Fighters Johnny Preston Coolio Shania Twain Mr Mister Ella Henderson Spandau Ballet Luther Vandross Barry White Paul Young Salt N Pepper M People Terence Trent D'arby Sea Horses Drifters Louis Armstrong Fun Boy Three Gene Pitney Monty Python Frank Sinatra Talking Heads Pointer Sisters Mika Britney Spears Chaka Khan Fugees Green Day Communards Steps Supergrass Arctic Monkeys Neil Diamond Copyright QOD Page 2/38 Team/Player Sheet 1 Game 1 Irene Cara Thin Lizzy -

Schedule Quickprint TKRN-FM
Schedule QuickPrint TKRN-FM 3/5/2021 7PM through 3/5/2021 11P s: AirTime s: Runtime Schedule: Description 07:00:00p 00:00 Friday, March 05, 2021 7PM 07:00:00p 02:48 DANCING WITH A STRANGER / SAM SMITH & NORMANI 07:02:48p 04:00 KARMA CHAMELEON / CULTURE CLUB 07:06:48p 03:23 I HOPE / GABBY BARRETT FEATURING CHARLIE PUTH 07:10:11p 03:11 BEST DAY OF MY LIFE / AMERICAN AUTHORS 07:13:22p 03:28 INTENTIONS / JUSTIN BIEBER FEATURING QUAVO 07:16:50p 03:29 NO SCRUBS / TLC 07:20:19p 03:38 BREAK MY HEART / DUA LIPA 07:23:57p 03:16 BYE BYE BYE / 'N SYNC 07:27:17p 03:30 STOP-SET 07:34:04p 03:12 THE BONES / MAREN MORRIS 07:37:16p 04:13 DROPS OF JUPITER (TELL ME) / TRAIN 07:41:29p 04:04 CRAZY FOR YOU / MADONNA 07:45:33p 03:09 MOOD (MIXSHOW EDIT CLEAN) / 24KGOLDN 07:48:42p 03:39 LOVE DON'T COST A THING / JENNIFER LOPEZ 07:52:21p 03:30 STOP-SET 08:00:00p 00:00 Friday, March 05, 2021 8PM 08:00:00p 02:58 TRAMPOLINE / SHAED 08:02:58p 03:54 EYE OF THE TIGER / SURVIVOR 08:06:52p 03:24 ADORE YOU / HARRY STYLES 08:10:16p 03:27 COME ON GET HIGHER / MATT NATHANSON 08:13:43p 03:02 MEMORIES / MAROON 5 08:16:45p 03:45 TOTAL ECLIPSE OF THE HEART / NICKI FRENCH 08:20:30p 03:28 CIRCLES / POST MALONE 08:23:58p 02:56 THUNDER / IMAGINE DRAGONS 08:26:58p 03:30 STOP-SET 08:33:45p 03:17 WITHOUT ME (CLEAN VERSION) / HALSEY 08:37:02p 03:54 STAY / RIHANNA FEATURING MIKKY EKKO 08:40:56p 04:22 I STILL HAVEN'T FOUND WHAT I'M LOOKING FOR / U2 08:45:18p 03:20 LEVITATING (RAP-LESS EDIT) / DUA LIPA 08:48:38p 03:50 SHAPE OF YOU / ED SHEERAN 08:52:28p 03:30 STOP-SET 09:00:00p 00:00 Friday, -

Power Syndicate
Beautiful Disaster 311 You Shook Me All Night Long AC/DC Walk This Way Aerosmith Would Alice In Chains Bring The Noise Anthrax/Public Enemy (1986) Show Me How To Live Audioslave Fight For Your Right Beastie Boys Paul Revere Beastie Boys The Metro Berlin White Wedding Billy Idol Everybody Wants You Billy Squier Paranoid Black Sabbath One Way or Another Blondie Burning For You Blue Oyster Cult Crazy Bitch Buckcherry Surrender Cheap Trick Clean My Wounds Corrosion of Conformity Butterfly Crazy Town Pour Some Sugar on Me Def Leppard Whip It Devo Down With The Sickness Disturbed Heavy Metal (Takin' a Ride) Don Felder Anything, Anything Dramarama Girls on Film Duran Duran Lose Yourself Eminem Sweet Dreams Eurythmics Say What You Will Fastway Paralyzer Finger Eleven I Ran Flock of Seagulls All Right Now Free Whatever Godsmack We're An American Band Grand Funk Railroad Brain Stew Green Day Mr. Brownstone Guns 'N' Roses Unsung Helmet Tonight Kool & The Gang Break Stuff Limp Bizkit One Step Closer Linkin Park My Own Worst Enemy Lit Simple Man Lynyrd Skynyrd The Beautiful People Marilyn Manson Symphony of Destruction Megadeth Sad But True Metallica No Parking on the Dance Floor Midnight Star This Is How We Do It Montel Jordan Kickstart My Heart Motley Crue Ace of Spades Motorhead Hella Good No Doubt Come Out And Play Offspring Hit Me With Your Best Shot Pat Benetar Snortin' Whiskey, Drinkin' Cocaine Pat Travers Band Young Lust Pink Floyd Metal Health Quiet Riot Killing In The Name Of Rage Against The Machine Sleep Now In The Fire Rage Against The