Crosstalk Between Histone Modifications Indicates That
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Searching for Inhibitors of the Protein Arginine Methyl Transferases: Synthesis and Characterisation of Peptidomimetic Ligands
SEARCHING FOR INHIBITORS OF THE PROTEIN ARGININE METHYL TRANSFERASES: SYNTHESIS AND CHARACTERISATION OF PEPTIDOMIMETIC LIGANDS by ASTRID KNUHTSEN B. Sc., Aarhus University, 2009 M. Sc., Aarhus University, 2012 A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Pharmaceutical Sciences) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) March 2016 © Astrid Knuhtsen, 2016 UNIVERSITY OF COPENH AGEN FACULTY OF HEALTH A ND MEDICAL SCIENCES PhD Thesis Astrid Knuhtsen Searching for Inhibitors of the Protein Arginine Methyl Transferases: Synthesis and Characterisation of Peptidomimetic Ligands December 2015 This thesis has been submitted to the Graduate School of The Faculty of Health and Medical Sciences, University of Copenhagen ii Thesis submission: 18th of December 2015 PhD defense: 11th of March 2016 Astrid Knuhtsen Department of Drug Design and Pharmacology Faculty of Health and Medical Sciences University of Copenhagen Universitetsparken 2 DK-2100 Copenhagen Denmark and Faculty of Pharmaceutical Sciences University of British Columbia 2405 Wesbrook Mall BC V6T 1Z3, Vancouver Canada Supervisors: Principal Supervisor: Associate Professor Jesper Langgaard Kristensen Department of Drug Design and Pharmacology, University of Copenhagen, Denmark Co-Supervisor: Associate Professor Daniel Sejer Pedersen Department of Drug Design and Pharmacology, University of Copenhagen, Denmark Co-Supervisor: Assistant Professor Adam Frankel Faculty of Pharmaceutical -

Asymmetric Expression of Lincget Biases Cell Fate in Two-Cell Mouse Embryos
UC Riverside UC Riverside Previously Published Works Title Asymmetric Expression of LincGET Biases Cell Fate in Two-Cell Mouse Embryos. Permalink https://escholarship.org/uc/item/3100h0cm Journal Cell, 175(7) ISSN 0092-8674 Authors Wang, Jiaqiang Wang, Leyun Feng, Guihai et al. Publication Date 2018-12-01 DOI 10.1016/j.cell.2018.11.039 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Article Asymmetric Expression of LincGET Biases Cell Fate in Two-Cell Mouse Embryos Graphical Abstract Authors Jiaqiang Wang, Leyun Wang, Guihai Feng, ..., Zhonghua Liu, Wei Li, Qi Zhou Correspondence [email protected] (W.L.), [email protected] (Q.Z.) In Brief An endogenous retrovirus-associated nuclear long noncoding RNA biases cell fate in mouse two-cell embryos. Highlights Data Resources d LincGET is asymmetrically expressed in the nucleus of two- GSE110419 to four-cell mouse embryos d LincGET overexpression biases blastomere fate toward inner cell mass (ICM) d LincGET physically binds to CARM1 d LincGET/CARM1 activates ICM-specific genes Wang et al., 2018, Cell 175, 1887–1901 December 13, 2018 ª 2018 Elsevier Inc. https://doi.org/10.1016/j.cell.2018.11.039 Article Asymmetric Expression of LincGET Biases Cell Fate in Two-Cell Mouse Embryos Jiaqiang Wang,1,2,3,4,8 Leyun Wang,1,8 Guihai Feng,1,8 Yukai Wang,1,8 Yufei Li,1,2 Xin Li,5,6 Chao Liu,1,2 Guanyi Jiao,1,2 Cheng Huang,1,2 Junchao Shi,7 Tong Zhou,7 Qi Chen,7 Zhonghua Liu,4 Wei Li,1,2,3,* and Qi Zhou1,2,3,9,* 1State Key Laboratory of Stem Cell and Reproductive -

Establishment and Optimization of the Chrop Approach, Combining Chip and MS-Based Proteomics
PhD degree in Molecular Medicine European School of Molecular Medicine (SEMM), University of Milan and University of Naples “Federico II” Faculty of Medicine Settore disciplinare: BIO/10 Establishment and optimization of the ChroP approach, combining ChIP and MS-based proteomics, for the characterization of the chromatome at distinct functional domains Monica Soldi IFOM-IEO Campus, Milan Matricola n. R08407 Supervisor: Dr. Tiziana Bonaldi IFOM-IEO Campus, Milan Added co-Supervisor: Dr. Gioacchino Natoli IFOM-IEO Campus, Milan Anno accademico 2011-2012 To my family: Andrea and Paolo! 1 TABLE OF CONTENT FIGURE AND TABLE INDEX ....................................................................................... 5 LIST OF ABBREVIATIONS ....................................................................................... 8 1. ABSTRACT .............................................................................................................. 10 2. INTRODUCTION .................................................................................................... 13 2.1. Chromatin, epigenetics and histone post-translational modifications ............... 13 2.2. Mass Spectrometry analysis and MS-based proteomics ...................................... 17 2.2.1. Basic concepts of Mass Spectrometry........................................................... 19 2.2.2. Different MS approaches for hPTMs analysis: from “Bottom Up” to “Top Down”, via “Middle Down” .................................................................................. 25 -

Supporting Information
Supporting Information Chu et al. 10.1073/pnas.1323226111 SI Materials and Methods phenylcarbamate, O-GlcNAz, and phosphine-FLAG were pur- Cell Culture. Human breast cancer cell line MCF7 was cultured in chased from Sigma. DMEM/F12 containing 10% (vol/vol) FBS (Sigma), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). Human kidney Plasmids. Human full-length OGT (NP_858058.1) was cloned fibroblast HEK 293T and breast cancer cell line MDA-MB-231 from 293T cDNA into pcDNA3.1-V5-HIS (Invitrogen). Human were cultured in DMEM (Gibco) containing 10% (vol/vol) FBS full-length EZH2 (NP_001190176.1) was cloned from 293T (Sigma), 100 U/mL penicillin, and 100 μg/mL streptomycin cDNA into pcDNA3-FLAG. Resistant OGT and EZH2-S75A were constructed using a site-directed mutagenesis method as (Gibco). All cells were cultured at 37 °C with 5% (vol/vol) CO2. described by the manufacturer (Finnzyme). The primers used for Knockdown by siRNA. Briefly, cells at 10% confluence in six-well construction of the resistant OGT plasmid against siOGT siRNA plates were transfected with 20 nM of siRNA by Lipofectamine are as follows: forward primer, 5′-CTA TGA ATA CTA TTG 2000 (Invitrogen) for 3 d. Cells were then harvested for further CGG AGG CGG TGA TCG AGA TGA TCA ACC GAG GAC use. The siRNAs used are Scramble siRNA (Invitrogen), siOGT#1 AGA T-3′ and reverse primer, 5′-ATC TGT CCT CGG TTG (sense, 5′-UAAUCAUUUCAAUAACUGCUUCUGC-3′,and ATC ATC TCG ATC ACC GCC TCC GCA ATA GTA TTC antisense, 5′-GCAGAAGCAGUUAUUGAAAUGAUUA-3′)and ATA G-3′. -
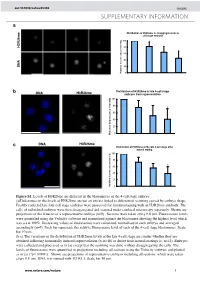
Histone Arginine Methylation Regulates Pluripotency in the Early
doi: 10.1038/nature05458 SUPPLEMENTARY INFORMATION a e Distribution of H3R26me in disaggregated late 4- cell stage embryos m 6 2 100 R 3 H 80 60 40 A N D 20 Relative fluorescence intensity 0 b Distribution of H3R26me in late 4-cell stage DNA H3R26me embryos from superovulation 100 80 * 60 40 20 0 Relative fluorescence intensity DNA H3R26me c Distribution of H3R26me at the late 4-cell stage after natural mating 100 80 60 * 40 20 Relative fluorescence intensity 0 Figure S1. Levels of H3R26me are different in the blastomeres of the 4-cell stage embryo. (a)Differences in the levels of H3R26me are not an artifact linked to differential scanning caused by embryo shape. Freshly collected late four cell stage embryos were processed for immunostaining with an H3R26me antibody. The cells of individual embryos were then disaggregated and scanned under confocal microscopy separately. Shown are projections of the 4 nuclei of a representative embryo (n=9) . Sections were taken every 0.8 µm. Fluorescence levels were quantified using the Volocity software and normalised against the blastomere showing the highest level which was set at 100%. Decreasing values of fluorescence were calculated, normalised in each embryo and averaged accordingly (n=9). Each bar represents the relative fluorescence level of each of the 4-cell stage blastomeres. Scale bar 10 µm. (b-c) The variations in the distribution of H3R26me levels at the late 4-cell stage are similar whether they are obtained following hormonally induced superovulation (b; n=18) or derive from natural matings (c; n=14). -

Epigenetic Regulation of Mouse Preimplantation Embryo Development
Available online at www.sciencedirect.com ScienceDirect Epigenetic regulation of mouse preimplantation embryo development 1,2,3,6 1,2,3,6 1,2,3,4,5 Xudong Fu , Chunxia Zhang and Yi Zhang After fertilization, mouse embryos go through preimplantation major waves. While the minor ZGA occurs in zygotes and development to give rise to blastocyst. Two key molecular early two-cell embryos where about one hundred of events, zygotic genome activation (ZGA) and the first cell zygotic genes are transcribed [2], the major ZGA mainly lineage specification, are essential for the process. Recent takes place in late two-cell embryos with thousands of advances in low-input epigenomics profiling techniques allow genes are actively transcribed [3]. After ZGA, the the analysis of these events at a molecular level, which revealed embryos go through a few cell cleavages before the first a critical role of epigenetic and chromatin reprogramming in cell lineage specification to generate trophectoderm (TE) ZGA and the first cell lineage specification. Additionally, the and inner cell mass (ICM). During the past several years, establishment of an in vitro embryonic stem cell (ESC) to two- great progress has been made in understanding the cell embryo-like conversion system have also contributed to molecular events, particularly in ZGA and the first cell the molecular understanding of preimplantation development. lineage specification, thanks to the development of low- In this review, we summarize recent advances in epigenetic input epigenomic profiling techniques as well as the regulation of mouse preimplantation development, point out establishment of an in vitro cell fate conversion model the remaining questions, and propose strategies to tackle for early embryos. -

PRDM14 Is a Unique Epigenetic Regulator Stabilizing Transcriptional Networks for Pluripotency
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Kwansei Gakuin University Repository PRDM14 Is a Unique Epigenetic Regulator Stabilizing Transcriptional Networks for Pluripotency journal or Frontiers in Cell and Developmental Biology publication title volume 6 page range 1-5 year 2018-02-13 URL http://hdl.handle.net/10236/00026967 MINI REVIEW published: 13 February 2018 doi: 10.3389/fcell.2018.00012 PRDM14 Is a Unique Epigenetic Regulator Stabilizing Transcriptional Networks for Pluripotency Yoshiyuki Seki* Department of Biomedical Chemistry, School of Science and Technology, Kwansei Gakuin University, Hyogo, Japan PR-domain containing protein 14 (PRDM14) is a site-specific DNA-binding protein and is required for establishment of pluripotency in embryonic stem cells (ESCs) and primordial germ cells (PGCs) in mice. DNA methylation status is regulated by the balance between de novo methylation and passive/active demethylation, and global DNA hypomethylation is closely associated with cellular pluripotency and totipotency. PRDM14 ensures hypomethylation in mouse ESCs and PGCs through two distinct layers, transcriptional repression of the DNA methyltransferases Dnmt3a/b/l and active demethylation by recruitment of TET proteins. However, the function of PRDM14 remains unclear in other species including humans. Hence, here we focus on the unique characteristics of mouse PRDM14 in the epigenetic regulation of pluripotent cells and primordial germ cells. In addition, we discuss the expression regulation and function of PRDM14 in other species Edited by: Alexey Ruzov, compared with those in mice. University of Nottingham, United Kingdom Keywords: Prdm14, epigenetics, embryonic stem cells (ESCs), primordial germ cells, DNA Methylation Reviewed by: Ian C. -

DNA Methylation Targeting: the DNMT/HMT Crosstalk Challenge
biomolecules Review DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge Omar Castillo-Aguilera 1, Patrick Depreux 1, Ludovic Halby 2, Paola B. Arimondo 2,3,* and Laurence Goossens 1,* 1 Univ. Lille, ICPAL, EA 7365—GRITA—Groupe de Recherche sur les formes Injectables et les Technologies Associées, 3 rue du Pr. Laguesse, F-59000 Lille, France; [email protected] (O.C.-A.); [email protected] (P.D.) 2 FRE3600 Epigenetic Targeting of Cancer, CNRS, 31035 Toulouse, France; [email protected] 3 Churchill College, Cambridge CB3 0DS, UK * Correspondence: [email protected] (P.B.A.); [email protected] (L.G.); Tel.: +33-5-61335919 (P.B.A.); +33-3-20-96-47-02 (L.G.) Academic Editors: Gerda Egger and Melanie R. Hassler Received: 15 November 2016; Accepted: 12 December 2016; Published: 5 January 2017 Abstract: Chromatin can adopt a decondensed state linked to gene transcription (euchromatin) and a condensed state linked to transcriptional repression (heterochromatin). These states are controlled by epigenetic modulators that are active on either the DNA or the histones and are tightly associated to each other. Methylation of both DNA and histones is involved in either the activation or silencing of genes and their crosstalk. Since DNA/histone methylation patterns are altered in cancers, molecules that target these modifications are interesting therapeutic tools. We present herein a vast panel of DNA methyltransferase inhibitors classified according to their mechanism, as well as selected histone methyltransferase inhibitors sharing a common mode of action. Keywords: DNA methylation; histone methylation; DNMT/HMT crosstalk; DNMT inhibitors; HMT inhibitors 1. -
Systems Level Analysis of Histone H3 Post-Translational Modifications (Ptms) Reveals Features of PTM Crosstalk in Chromatin Regulation
University of Southern Denmark Systems Level Analysis of Histone H3 Post-translational Modifications (PTMs) Reveals Features of PTM Crosstalk in Chromatin Regulation Schwämmle, Veit; Sidoli, Simone; Ruminowicz, Chrystian; Wu, Xudong; Lee, Chung-Fan; Helin, Kristian; Jensen, Ole Nørregaard Published in: Molecular and Cellular Proteomics DOI: 10.1074/mcp.M115.054460 Publication date: 2016 Document version: Final published version Citation for pulished version (APA): Schwämmle, V., Sidoli, S., Ruminowicz, C., Wu, X., Lee, C-F., Helin, K., & Jensen, O. N. (2016). Systems Level Analysis of Histone H3 Post-translational Modifications (PTMs) Reveals Features of PTM Crosstalk in Chromatin Regulation. Molecular and Cellular Proteomics, 15(8), 2715-2729. https://doi.org/10.1074/mcp.M115.054460 Go to publication entry in University of Southern Denmark's Research Portal Terms of use This work is brought to you by the University of Southern Denmark. Unless otherwise specified it has been shared according to the terms for self-archiving. If no other license is stated, these terms apply: • You may download this work for personal use only. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying this open access version If you believe that this document breaches copyright please contact us providing details and we will investigate your claim. Please direct all enquiries to [email protected] Download date: 09. Oct. 2021 crossmark Research © 2016 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Systems Level Analysis of Histone H3 Post- translational Modifications (PTMs) Reveals Features of PTM Crosstalk in Chromatin Regulation*□S Veit Schwa¨mmle‡ʈ**, Simone Sidoli‡§§**, Chrystian Ruminowicz‡, Xudong Wu§‡‡, Chung-Fan Lee§, Kristian Helin§¶, and Ole N. -

Symmetric-Di-Methyl-Histone H3 (Arg17) Rabbit Pab
Symmetric-Di-methyl-Histone H3 (Arg17) rabbit pAb (H3R17me2(sy) pAb) www.ptm-biolab.com.cn Cat#: PTM-679 [email protected] Species: Rabbit 0571-2883 3567 Size: 100 μl Form: supplied in liquid form Recommend Species Swissprot Molecular Application Immunogen dilution Reactivity ID Weight H3R17me2(sy) WB, Dot, ELISA 1:2000 for WB H, M, R P68431 17 KDa -KLH **Species reactivity is determined by WB. Kept at -20oC after reconstituted. *** Anti-rabbit secondary antibodies must be used to detect this antibody. Source/Purification: This product is produced by immunizing rabbits with a synthetic di-methyl(sy) peptide corresponding to residues surrounding Arg17 of human histone H3. Antibodies are purified by protein A-conjugated agarose followed by di-methylated(sy) histone H3 (Arg17) peptide affinity chromatography. Recommended Applications: ELISA, Dot blot, Western blot. Recommended antibody dilution: WB: 1:2000 NOTE: For WB, incubate membrane with diluted antibody in 5% nonfat milk, 1 x TBS, 0.1% Tween- 20 for two hours at room temperature with gentle shaking. Prepare working dilution immediately before use. Use at an assay dependent concentration. Optimal dilutions/concentrations should be determined by the end user. Not yet tested in other applications Scientific Description: Protein post-translational Modification (PTM) is an important reversible event controlling protein activity. The amino-terminal tails of core histones undergo multiple modifications in multiple sites, termed as “histone code” or “epigenetic code”. Arginine methylation in core histones is a major determinant for the formation of active and inactive regions of the genome and therefore plays vital roles in multiple cellular events. -

Anti-Mono-Methyl-Histone H3 (Arg42) Rabbit Pab
Anti-Mono-Methyl-Histone H3 (Arg42) Rabbit pAb Catalog # PTM-610 General Information Images Host species Rabbit Dot Blot Clonality Polyclonal Blocking buffer: 5% NFDM/TBST Primary ab dilution: 1:2000 Clone number / Primary ab incubation condition: 2 hours at room temperature Synonym H3R42me1 Secondary ab: Goat Anti-Rabbit IgG H&L (HRP) Immunogen UniProt MW (kDa) Applications Immunogen peptide quantity: 1 ng, 4 ng, 16 ng species Exposure time: 60 s Human P68431 15 WB The list of peptides is included in the table below: No. Peptide No. Peptide Product Usage Information Lane 1 H3R42me1 Lane 2 H3R17me1 Lane 3 H3R17me2 (sy) Lane 4 H3R17me2 (asy) Application Dilution Recommended species Lane 5 H3R17cit Lane 6 H3R26me1 WB 1:500-1:2000 Human, Mouse, Rat Lane 7 H3R26me2 (sy) Lane 8 H3R26me2 (asy) Lane 9 H3R26cit Lane 10 H3R42me2 (asy) Properties Lane 11 H3R42un Storage Store at -20 °C. Avoid freeze / thaw cycles. WB Stability Stable for 12 months from date of receipt / reconstitution Blocking buffer: 5% NFDM/TBST Primary ab dilution: 1:2000 Constituents PBS, Glycerol, BSA Primary ab incubation condition: 2 hours at Purity Antibodies are purified by Protein A conjugated agarose room temperature followed by Mono-methylated H3 (Arg42) peptide affinity Secondary ab: Goat Anti-Rabbit IgG H&L (HRP) Lysate: MCF-7, C2C12, N2a chromatography. Protein loading quantity: 20 μg Exposure time: 60 s Isotype IgG Predicted MW: 15 kDa Observed MW: 15 kDa Target Information Function Histone post-translational modifications (PTMs) are key Blocking buffer: 5% NFDM/TBST mechanisms of epigenetics that modulate chromatin Primary ab dilution: 1:2000 structures, termed as “histone code”. -

Molecular Marks for Epigenetic Identification of Developmental and Cancer Stem Cells
Clin Epigenet (2011) 2:27–53 DOI 10.1007/s13148-010-0016-0 REVIEW Molecular marks for epigenetic identification of developmental and cancer stem cells Samir Kumar Patra & Moonmoon Deb & Aditi Patra Received: 1 August 2010 /Accepted: 24 November 2010 /Published online: 17 December 2010 # Springer-Verlag 2010 Abstract Epigenetic regulations of genes by reversible through retroviral techniques. In this contribution, we will methylation of DNA (at the carbon-5 of cytosine) and discuss some of the major discoveries on epigenetic mod- numerous reversible modifications of histones play important ifications within the chromatin of various genes associated roles in normal physiology and development, and epigenetic with cancer progression and cancer stem cells in comparison deregulations are associated with developmental disorders and to normal development of stem cell. These modifications may various disease states, including cancer. Stem cells have the be considered as molecular signatures for predicting disorders capacity to self-renew indefinitely. Similar to stem cells, some of development and for identifying disease states. malignant cells have the capacity to divide indefinitely and are referred to as cancer stem cells. In recent times, direct Keywords Cancer . Development . DNA-methylation . correlation between epigenetic modifications and reprogram- Histone modification . Epigenetics . Cancer Stem cell ming of stem cell and cancer stem cell is emerging. Major discoveries were made with investigations on reprogramming gene products, also known as master regulators of totipotency Introduction and inducer of pluoripotency, namely, OCT4, NANOG, cMYC, SOX2, Klf4, and LIN28. The challenge to induce Defining the growth requirements for the maintenance pluripotency is the insertion of four reprogramming genes and differentiation of developmental and cancer cells, (Oct4, Sox2, Klf4, and c-Myc) into the genome.