[Thesis Title Goes Here]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Invention of the Transistor
The invention of the transistor Michael Riordan Department of Physics, University of California, Santa Cruz, California 95064 Lillian Hoddeson Department of History, University of Illinois, Urbana, Illinois 61801 Conyers Herring Department of Applied Physics, Stanford University, Stanford, California 94305 [S0034-6861(99)00302-5] Arguably the most important invention of the past standing of solid-state physics. We conclude with an century, the transistor is often cited as the exemplar of analysis of the impact of this breakthrough upon the how scientific research can lead to useful commercial discipline itself. products. Emerging in 1947 from a Bell Telephone Laboratories program of basic research on the physics I. PRELIMINARY INVESTIGATIONS of solids, it began to replace vacuum tubes in the 1950s and eventually spawned the integrated circuit and The quantum theory of solids was fairly well estab- microprocessor—the heart of a semiconductor industry lished by the mid-1930s, when semiconductors began to now generating annual sales of more than $150 billion. be of interest to industrial scientists seeking solid-state These solid-state electronic devices are what have put alternatives to vacuum-tube amplifiers and electrome- computers in our laps and on desktops and permitted chanical relays. Based on the work of Felix Bloch, Ru- them to communicate with each other over telephone dolf Peierls, and Alan Wilson, there was an established networks around the globe. The transistor has aptly understanding of the band structure of electron energies been called the ‘‘nerve cell’’ of the Information Age. in ideal crystals (Hoddeson, Baym, and Eckert, 1987; Actually the history of this invention is far more in- Hoddeson et al., 1992). -

A Pictorial History of Nuclear Instrumentation
A Pictorial History of Nuclear Instrumentation Ranjan Kumar Bhowmik Inter University Accelerator Centre (Retd) Dawn of Nuclear Instrumentation End of 19th Century Wilhelm Röntgen (1845-1923) (1895) W. Roentgen observes florescence in barium platinocyanide due to invisible rays from a gas discharge tube. Names them X- rays First medical X-ray Henry Becquerel (1852-1908) (1896) H. Bacquerel detects that uranium salts spontaneously emit a penetrating radiation that can blacken photographic films Independent of chemical composition Pierre Curie (1859-1906) Marie Curie (1867-1934) (1987) Pierre & Marie Curie found that both uranium and thorium emit radiation that can ionise gases – detected by electroscopes. Coin the name ‘Radioactivity’ to describe this natural process Early Instrumentation Equipment used for these path-breaking discoveries were developed much earlier : • Optical fluorescence (1560) Bernardino de Sahagún • Thermo-luminescence (17th Century) • Gold Leaf Electroscope (1787) Abraham Bennet • Photographic Emulsion (1839) Louis Daguerre Early instruments were sensitive to radiation dose only, could not detect individual radiation Spinthariscope (1903) • Spinthariscope was invented by William Crookes. It is made of a ZnS screen viewed by an eyepiece • Scintillation produced by an incident a-particle can be seen as faint light flashes • Rutherford’s famous a-scattering experiment proved the existence a ‘point-like’ nucleus inside the atom Geiger Counter (1908) First instrument to detect individual a-particles electronically was developed by Geiger. Chamber filled with CO2 at 2-5 cm of mercury Central wire at ground potential connected to an electrometer. Successive ‘kicks’ in the electrometer indicated the passage of a charged particle; response time ~1 sec Geiger Muller Counter (1928) An improved design (1912) had a string Major improvement was electrometer for detection. -

The General Working of Solar Cells and the Correlation Between
The general working of solar cells and the correlation between diffuseness and temperature, irradiance and spectral shape Thomas Kalkman & Max Verweg Under supervision of M. Futscher and B. Ehrler 2 week Bachelor Research Project June 28, 2017 Physics and Astronomy, University of Amsterdam [email protected], [email protected] Abstract main parameter for efficiency would bring clarification and demands for further experiments to verify this parameter. The efficiency of solar cells is mostly measured under standard test conditions. In reality, the temperature and Theory sunlight is not always the same. In The Netherlands there is on average only 1650 hours of sunshine per year[2], the Solar cells are made of different layers of materials. They rest of the daytime per year is without direct sunlight ra- have a protective glass plate, thin films as moisture barriers diating the solar cells. The light captured is diffuse. In this and the actual solar cells which convert the energy. work we discuss the basics of how solar cells work and we investigate the correlation between diffuseness of the sunlight and temperature, irradiation and spectral shape. We find correlations due to weather conditions. Further- more we verify correlations between efficiency and tem- perature, and efficiency and irradiation. Introduction Figure 1: Schematic representation of a silicon solar cell. Since the discovery of the photovoltaic effect - explaining The blue layer is an anti reflective and protective layer, the how electricity can be converted from sunlight - by Alexan- grey layers are the cathode and anode, the yellow layers are dre Edmond Becquerel in 1839, and the invention of solar the silicon semiconductor. -

Solar Cells Operation and Modeling
Solar Cells Operation and Modeling Dragica Vasileska, ASU Gerhard Klimeck, Purdue Outline – Introduction to Solar Cells – Absorbing Solar Energy – Solar Cell Equations Solution • PV device characteristics • Important generation-recombination mechanisms • Analytical model • Numerical Solution • Shadowing • Photon recycling • Quantum efficiency for current collection • Simulation of Solar Cells with Silvaco Introduction to Solar Cells - Historical Developments - o 1839: Photovoltaic effect was first recognized by French physicist Alexandre-Edmond Becquerel. o 1883: First solar cell was built by Charles Fritts, who coated the semiconductor selenium with an extremely thin layer of gold to form the junctions (1% efficient). o 1946: Russell Ohl patented the modern solar cell o 1954: Modern age of solar power technology arrives - Bell Laboratories, experimenting with semiconductors, accidentally found that silicon doped with certain impurities was very sensitive to light. o The solar cell or photovoltaic cell fulfills two fundamental functions: o Photogeneration of charge carriers (electrons and holes) in a light- absorbing material o Separation of the charge carriers to a conductive contact to transmit electricity Introduction to Solar Cells - Solar Cells Types - • Homojunction Device - Single material altered so that one side is p-type and the other side is n-type. - p-n junction is located so that the maximum amount of light is absorbed near it. • Heterojunction Device - Junction is formed by contacting two different semiconductor. - Top layer - high bandgap selected for its transparency to light. - Bottom layer - low bandgap that readily absorbs light. • p-i-n and n-i-p Devices - A three-layer sandwich is created, - Contains a middle intrinsic layer between n-type layer and p-type layer. -

Impact of Bifacial Pv System on Canary Islands' Energy
IMPACT OF BIFACIAL PV SYSTEM ON CANARY ISLANDS’ ENERGY 4 DE SEPTIEMBRE DE 2018 MÁSTER DE ENERGÍAS RENOVABLES Universidad de La Laguna Alberto Mora Sánchez / Impact of Bifacial PV System on Canary Island’s Energy IMPACT OF BIFACIAL PV SYSTEM ON CANARY ISLANDS’ ENERGY Alberto Mora Sánchez, Máster de Energías Renovables, Universidad de La Laguna, Ricardo Guerrero Lemus, Departamento de Física e Instituto de Materiales y Nanotecnología, Universidad de La Laguna ABSTRACT This project proposes an investigation motivated by the interest in obtaining a better understanding of the role that the bifacial technology could play in a favorable environment such as the Canarias Islands. In order to develop a scientific knowledge about this technology and to be able to evaluate the functioning of a bifacial system with different configurations that can give important results and show that this type of installations could be fruitful in the near future as already foreseen in the reports of the International Technology Roadmap for Photovoltaic where they predict that will increase their shear in photovoltaics the next years. Bifacial Technology has the advantage of being able to use the back of the module to produce energy using diffuse and albedo radiations. During the project, the mini-modules will be manufactured and characterized, and then placed in out-door structures where measurements will be carried out. In addition, it will try to use the down shifter technique that improves the spectral response thanks to the displacement of wavelengths that are not absorbed by the cells. The main advantage of the bifacial modules is to take advantage of the albedo that reaches its rear side, so that, in this study different configurations will be studied in order to optimize the generation of energy for the specific environmental conditions of the location. -

The Transistor Revolution (Part 1)
Feature by Dr Bruce Taylor HB9ANY l E-mail: [email protected] t Christmas 1938, working in their small rented garage in Palo Alto, California, two The Transistor enterprising young men Acalled Bill Hewlett and Dave Packard finished designing a novel wide-range Wien bridge VFO. They took pictures of the instrument sitting on the mantelpiece Revolution (Part 1) in their house, made 25 sales brochures and sent them to potential customers. Thus began the electronics company Dr Bruce Taylor HB9ANY describes the invention of the that by 1995 employed over 100,000 people worldwide and generated annual tiny device that changed the course of radio history. sales of $31 billion. The oscillator used fve thermionic valves, the active devices that had been the mainstay of wireless communications for over 25 years. But less than a decade after HP’s frst product went on sale, two engineers working on the other side of the continent at Murray Hill, New Jersey, made an invention that was destined to eclipse the valve and change wireless and electronics forever. On Decem- ber 23rd 1947, John Bardeen and Walter Brattain at Bell Telephone Laboratories (the research arm of AT&T) succeeded in making the device that set in motion a technological revolution beyond their wildest dreams. It consisted of two gold contacts pressed on a pinhead of semi-conductive material on a metallic base. The regular News of Radio item in the 1948 New York Times was far from being a blockbuster column. Relegated to page 46, a short article in the edition of July 1st reported that CBS would be starting two new shows for the summer season, “Mr Tutt” and “Our Miss Brooks”, and that “Waltz Time” would be broadcast for a full hour on three successive Fridays. -

Crystal Fire: the Birth of the Information Age
Crystal Fire: The Birth of the Information Age Theme: Sources Harold Fox “Silicon” is a compound that defines much of the modern technological era. The term connotes high-technology, exponential progress in Moore’s law, Silicon Valley, solar panels, even plastic surgery. The source of this transformation in large part arises from a single invention, the transistor, a sandwich of different types of silicon crystals. “Crystal Fire,” by Lillian Hoddeson and Michael Riordan, describes the invention of this device and its development up to the beginning of the microelectronic age of the 1960s. This is an unusually compelling story, both because of the invention’s huge impact and because of its straightforward progression of events involving a small number of people. The authors do an excellent job of telling the story as well as setting the institutional, scientific, and personal context. The history of computing occupies a unique position in the history of technology. Because of its rapid changes and near-instant impact on society, there are complete stories to tell now. Intriguingly, many of the people involved in the critical events are still alive, permitting the genesis of new ideas and products to be followed in great detail. There is also a wealth of documentation, easily accessible. At the same time, it is more difficult to get a perspective of objective distance. People and companies have reputations to sustain, and they will work to steer the narrative in their direction. Historians must be savvy journalists. Hoddeson and Riordan do a good job with their sources. Hoddeson herself had been working on solid-state physics history for 25 years. -

Pioneers of Computing
Social and Professional Issues in IT Pioneers of Computing Pioneers of Computing Many scientist and engineers have contributed in the development of computing science. This chapter covers all the major scientists and their works, who have played a significant role in the development of computing. Some of the major Pioneers of Computing and their contributions are described here Aristotle (384 BC, Stagira, Greece, 322 BC, Athens, Greece) He was one of the greatest ancient Greek philosophers of his time. Many of his thoughts have become the back bone of computing and artificial Intelligence. His work in the natural and social sciences greatly influenced virtually every area of modern thinking. Biography Aristotle was born in 384 BC in Stagira, on the northwest coast of the Aegean Sea. His father was a friend and the physician of the king of Macedonia, and the lad spent most of his boyhood at the court. At 17, he went to Athens to study. He enrolled at the famous Academy directed by the philosopher Plato. Aristotle threw himself wholeheartedly into Plato's pursuit of truth and goodness. Plato was soon calling him the "mind of the school." Aristotle stayed at the Academy for 20 years, leaving only when his beloved master died in 347 BC. In later years he renounced some of Plato's theories and went far beyond him in breadth of knowledge. Aristotle became a teacher in a school on the coast of Asia Minor. He spent two years studying marine biology on Lesbos. In 342 BC, Philip II invited Aristotle to return to the Macedonian court and teach his 13-year-old son Alexander. -

Evolution of High Effiency Silicon Solar Cell Design: Lecture 1
Australian Centre for Advanced Photovoltaics “Evolution of High Efficiency Silicon Solar Cell Design” Martin A. Green University of New South Wales UNSW Photovoltaics - Electricity from Sunlight Outline –Lecture I 1. Recent developments 2. Early PV history 3. The first pn-junction 4. Conventional space cells 5. Key pointers pn junctions 6. Enter the modern era -Questions- UNSW Photovoltaics - Electricity from Sunlight Annual capacity increase Wind 45 36 Gas Turbines New Capacity, GW New Capacity, . 27 18 Photovoltaics 9 0 Nuclear 2005 2006 2007 2008 2009 2010 2011 2012 UNSW PhotovoltaicsSources: - Electricity EPVIA, from SunlightIAEA, GWEA Ultimate potential? 2003 50.7 Terawatt UNSW Photovoltaics - Electricity from Sunlight “Submerged” progress 100000 25% 10000 World’s energy 1000 Nuclear Wind 100 Installed capacity, GW WBGU Scenario 1% milestone 1% World’s Projected Wind Actual electricity WBGU PV 10 PV Projected Actual PV 1% milestone PV WBGU Nuclear Actual Nuclear 1 2000 2010 2020 2030 2040 2050 UNSW Photovoltaics - Electricity from Sunlight German grid : May 2012 Actual production DE 30GW wind; 25GW PV 5/12 Peak demand 68GW (30% solar) displayed month: May 2012 MW 60,000 50,000 40,000 30,000 20,000 10,000 Peak conventional 51.2GW Tu We Th Fr Sa Su Mo Tu We Th Fr Sa Su Mo Tu We Th Fr Sa Su Mo Tu We Th Fr Sa Su Mo Tu We Th 01 02 03 04 05 06 07 08 09 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 Legend: Conventional > 100 MW Wind Solar UNSW Photovoltaics - Electricity from Sunlight Outline –Lecture I 1. -

Transistorized!, the One Hour Documentary Airing on PBS, Tells the Compelling Story of the History of the Transistor and the Scientists Who Discovered It
About the PBS Documentary The transistor is one of the 20th century’s most important inventions. It revolutionized technology and launched the Information Age. Its creation is a dramatic story of top secret research, serendipitous accidents, collaborative genius and clashing egos. Transistorized!, the one hour documentary airing on PBS, tells the compelling story of the history of the transistor and the scientists who discovered it. They include William Shockley, who assembled the team at Bell Labs that built the first working transistors, but whose driving ego ultimately ended their collaboration; John Bardeen, a theoretical genius whose profound insights paved the way to the final discovery; and Walter Brattain, whose persistent tinkering led to the breakthrough that resulted in the first transistor. Host Ira Flatow leads us through a vivid and Host Ira Flatow takes viewers entertaining tour of the key moments in the history back in time to recapture the of the transistor — from the scientific excitement and drama breakthroughs early in the 20th century that behind the invention that set the stage for the invention, through the changed the world – the Transistorized! transistor — in the PBS frustrations and serendipitous accidents that program “Transistorized!” will be broadcast made the first transistor work, to the evolution of the first transistorized products nationally on PBS and the birth of Silicon Valley. All inextricably interwoven with the tale of the brilliant Monday, November 8, collaboration and dramatic demise of the team that made the transistor possible. 1999 at 10pm ET. (Check local listings for the broadcast To learn more about the transistor visit www.pbs.org/transistor. -

The Solid State Diode
13-i 2/4/2016 Experiment 13 THE SOLID STATE DIODE INTRODUCTION – THE PN JUNCTION DIODE 1 THEORY – THE DIODE EQUATION 2 EXPERIMENTAL SETUP 4 EXPERIMENTAL PROCEDURE 6 Initial diode sweeps ................................................................................................................... 7 Calibration ................................................................................................................................. 8 Calibrated data collection ....................................................................................................... 12 Additional observations .......................................................................................................... 12 ANALYSIS 13 PRELAB PROBLEMS 14 APPENDIX – ELEMENTARY PHYSICS OF THE PN JUNCTION DIODE 15 Insulators, conductors, and semiconductors........................................................................... 15 Electrons and holes; impurities and doping ............................................................................ 17 The equilibrium PN junction .................................................................................................... 20 The PN junction I-V characteristic curve ................................................................................. 21 Zener and avalanche breakdown ............................................................................................ 24 Thermal behavior of the charge carriers ................................................................................. 26 REFERENCES: -
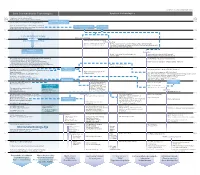
Download PDF File
Copyright©2017 Tokyo Electron Limited, All Right Reserved. Core Semiconductor Technologies Applied technologies 1876 Telephone invented by Graham Bell 1876 1897 Cathode ray tube invented by Karl Ferdinand Braun 1897 1900 Telegraph, telephone, 1900 Two-electrode vacuum tube invented by John Fleming wireless communication Three-electrode vacuum tube invented by Lee De Forest Wireless communication Vacuum tube Television receiver with a cathode ray tube devised technology technology by Russian scientist Boris Rosing 1910 1910 Semiconductor prehistory: Radio broadcasting 1920 Development of electronic circuit 1920 and control technology First radio station starts broadcasting TV broadcasting Radio broadcasting starts in the UK World’s first successful reception of images using a cathode ray tube Radio broadcasting starts in Japan Successful TV transmission experiment between New York and Washington DC Experimental TV broadcasting starts 1930 1930 Demand for durable solid state devices Computer Worlds’ first regular TV broadcasting starts Atanasoff‒Berry computer (ABC) invented in the UK (BBC) Bell Labs Model 1 relay computer introduced 1940 1940 Photovoltaic effect in silicon discovered by Russell Ohl (at Bell Labs) Codebreaking computer Colossus introduced P- and n-type conduction discovered by Jack Scaff Technique to manufacture p- and n-type semiconductors by World’s first general-purpose computer ENIAC completed doping impurities discovered by Henry Theuerer and Jack Scaff Point-contact transistor discovered by Walter Brattain and John Bardeen