Calculating Heat of Formation Values of Energetic Compounds: a Comparative Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hazardous Materials Table Quirements
Pipeline and Hazardous Materials Safety Admin., DOT § 172.101 Subpart H—Training this part, that person shall perform the function in accordance with this part. 172.700 Purpose and scope. 172.701 Federal-State relationship. [Amdt. 172–29, 41 FR 15996, Apr. 15, 1976, as 172.702 Applicability and responsibility for amended by Amdt. 172–32, 41 FR 38179, Sept. training and testing. 9, 1976] 172.704 Training requirements. Subpart B—Table of Hazardous Subpart I—Safety and Security Plans Materials and Special Provisions 172.800 Purpose and applicability. § 172.101 Purpose and use of haz- 172.802 Components of a security plan. ardous materials table. 172.804 Relationship to other Federal re- (a) The Hazardous Materials Table quirements. (Table) in this section designates the 172.820 Additional planning requirements for transportation by rail. materials listed therein as hazardous 172.822 Limitation on actions by states, materials for the purpose of transpor- local governments, and Indian tribes. tation of those materials. For each listed material, the Table identifies the APPENDIX A TO PART 172—OFFICE OF HAZ- hazard class or specifies that the mate- ARDOUS MATERIALS TRANSPORTATION rial is forbidden in transportation, and COLOR TOLERANCE CHARTS AND TABLES gives the proper shipping name or di- APPENDIX B TO PART 172—TREFOIL SYMBOL rects the user to the preferred proper APPENDIX C TO PART 172—DIMENSIONAL SPEC- IFICATIONS FOR RECOMMENDED PLACARD shipping name. In addition, the Table HOLDER specifies or references requirements in APPENDIX D TO PART 172—RAIL RISK ANAL- this subchapter pertaining to labeling, YSIS FACTORS packaging, quantity limits aboard air- craft and stowage of hazardous mate- AUTHORITY: 49 U.S.C. -

Organic Chemistry of Explosives
JWBK121-FM October 11, 2006 21:10 Char Count= 0 Organic Chemistry of Explosives Dr. Jai Prakash Agrawal CChem FRSC (UK) Former Director of Materials Defence R&D Organisation DRDO House, New Delhi, India email: [email protected] Dr. Robert Dale Hodgson Consultant Organic Chemist, Syntech Chemical Consultancy, Morecambe, Lancashire, UK Website: http://www.syntechconsultancy.co.uk email: [email protected] iii JWBK121-FM October 11, 2006 21:10 Char Count= 0 iii JWBK121-FM October 11, 2006 21:10 Char Count= 0 Organic Chemistry of Explosives i JWBK121-FM October 11, 2006 21:10 Char Count= 0 ii JWBK121-FM October 11, 2006 21:10 Char Count= 0 Organic Chemistry of Explosives Dr. Jai Prakash Agrawal CChem FRSC (UK) Former Director of Materials Defence R&D Organisation DRDO House, New Delhi, India email: [email protected] Dr. Robert Dale Hodgson Consultant Organic Chemist, Syntech Chemical Consultancy, Morecambe, Lancashire, UK Website: http://www.syntechconsultancy.co.uk email: [email protected] iii JWBK121-FM October 11, 2006 21:10 Char Count= 0 Copyright C 2007 John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England Telephone (+44) 1243 779777 Email (for orders and customer service enquiries): [email protected] Visit our Home Page on www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1T 4LP, UK, without the permission in writing of the Publisher. -

UPS Chemical Table
UPS Chemical Table - 49 CFR Version (Ground and Air Packages) BASIC DESCRIPTION FOR GROUND AND AIR GROUND AND AIR HAZARDOUS MATERIALS SHIPMENTS GROUND SHIPMENTS AIR SHIPMENTS SHIPMENTS Symbols: "‡" Requires a Technical Name / "*" For Ltd Qty see 49 CFR Part 173.*** / "**" See 49 CFR 173.27 for inner receptacle requirements / "▲" Accepted as CAO if amount is listed / "♦" PG = "II" or blank / "♦♦" PG = "III" or blank / "#" Sub-risk may be omitted from shipping papers if 49 CFR 172.400a(c) criteria met. (ref 172.202(a)(3)) CARGO HAZARD DOT PASSENGER AIRCRAFT DOT CLASS OR EXEMPTION, GROUND LTD QTY AIRCRAFT MAX NET EXEMPTION, HAZARDOUS MATERIALS DESCRIPTIONS DIVISION I.D. NUMBER LABEL(S) REQUIRED OR SPECIAL SERVICE TO LABEL(S) REQUIRED OR MAX NET MAX NET PER SPECIAL NON-BULK AND PROPER SHIPPING NAME (Subsidiary if (ALSO MARK PACKING EXEMPTION, SPECIAL PERMIT OR PERMIT CANADA EXEMPTION, SPECIAL PERMIT OR PER PER PACKAGE PERMIT SPECIAL EXCEPTIONS PACKAGING (ALSO MARK ON PACKAGE) applicable) ON PACKAGE) GROUP EXCEPTION OR 173.13 PERMITTED EXCEPTION PACKAGE** PACKAGE** ▲ OR 173.13 PROVISIONS §173.*** §173.*** (1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) Accellerene, see p-Nitrosodimethylaniline Accumulators, electric, see Batteries, wet etc Accumulators, pressurized, pneumatic or hydraulic (containing non-flamable gas), see Articles pressurized, pneumatic or hydraulic (containing non-flamable gas) Accumulators, pressurized, pneumatic or hydraulic (containing non-flammable gas), see Articles pressurized, pneumatic or hydraulic -

132 Subpart A—General Subpart B—Table of Hazardous Materials
§ 172.1 49 CFR Ch. I (10–1–12 Edition) 172.606 Carrier information contact. this part, that person shall perform the function in accordance with this part. Subpart H—Training [Amdt. 172–29, 41 FR 15996, Apr. 15, 1976, as 172.700 Purpose and scope. amended by Amdt. 172–32, 41 FR 38179, Sept. 172.701 Federal-State relationship. 9, 1976] 172.702 Applicability and responsibility for training and testing. Subpart B—Table of Hazardous 172.704 Training requirements. Materials and Special Provisions Subpart I—Safety and Security Plans § 172.101 Purpose and use of haz- 172.800 Purpose and applicability. ardous materials table. 172.802 Components of a security plan. (a) The Hazardous Materials Table 172.804 Relationship to other Federal re- (Table) in this section designates the quirements. materials listed therein as hazardous 172.820 Additional planning requirements materials for the purpose of transpor- for transportation by rail. tation of those materials. For each 172.822 Limitation on actions by states, listed material, the Table identifies the local governments, and Indian tribes. hazard class or specifies that the mate- APPENDIX A TO PART 172—OFFICE OF HAZ- rial is forbidden in transportation, and ARDOUS MATERIALS TRANSPORTATION gives the proper shipping name or di- COLOR TOLERANCE CHARTS AND TABLES rects the user to the preferred proper APPENDIX B TO PART 172—TREFOIL SYMBOL shipping name. In addition, the Table APPENDIX C TO PART 172—DIMENSIONAL SPEC- IFICATIONS FOR RECOMMENDED PLACARD specifies or references requirements in HOLDER this subchapter pertaining to labeling, APPENDIX D TO PART 172—RAIL RISK ANAL- packaging, quantity limits aboard air- YSIS FACTORS craft and stowage of hazardous mate- rials aboard vessels. -
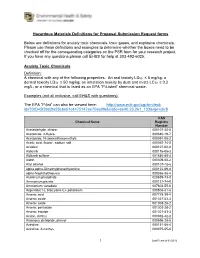
Hazardous Materials Definitions for Proposal Submission Request Forms
Hazardous Materials Definitions for Proposal Submission Request forms Below are definitions for acutely toxic chemicals, toxic gases, and explosive chemicals. Please use these definitions and examples to determine whether the boxes need to be checked off for the corresponding categories on the PSR form for your research project. If you have any questions please call EH&S for help at 303-492-6025. Acutely Toxic Chemicals Definition: A chemical with any of the following properties: An oral toxicity LD50 ≤ 5 mg/kg; a dermal toxicity LD50 ≤ 50 mg/kg; an inhalation toxicity by dust and mists LC50 ≤ 0.2 mg/L; or a chemical that is listed as an EPA “P-Listed” chemical waste. Examples (not all inclusive, call EH&S with questions): The EPA “P-list” can also be viewed here: http://www.ecfr.gov/cgi-bin/text- idx?SID=5f26d2fa35cbe51c4475147ee70aa0f6&node=se40.26.261_133&rgn=div8 CAS Chemical Name Registry Number Acetaldehyde, chloro- 000107-20-0 Acetamide, 2-fluoro- 000640-19-7 Acetamide, N-(aminothioxomethyl)- 000591-08-2 Acetic acid, fluoro-, sodium salt 000062-74-8 Acrolein 000107-02-8 Aldicarb 000116-06-3 Aldicarb sulfone 001646-88-4 Aldrin 000309-00-2 Allyl alcohol 000107-18-6 alpha,alpha-Dimethylphenethylamine 000122-09-8 alpha-Naphthylthiourea 000086-88-4 Aluminum phosphide 020859-73-8 Ammonium picrate 000131-74-8 Ammonium vanadate 007803-55-6 Argentate(1-), bis(cyano-C)-,potassium 000506-61-6 Arsenic acid 007778-39-4 Arsenic oxide 001327-53-3 Arsenic oxide 001303-28-2 Arsenic pentoxide 001303-28-2 Arsenic trioxide 001327-53-3 Arsine, diethyl 000692-42-2 Arsonous dichloride, phenyl- 000696-28-6 Aziridine 000151-56-4 Aziridine, 2-methyl- 000075-55-8 1 Last Revised 01/2016 Barium cyanide 000542-62-1 Benzenamine, 4-chloro- 000106-47-8 Benzenamine, 4-nitro- 000100-01-6 Benzene, (chloromethyl)- 000100-44-7 Benzeneethanamine, alpha,alpha- dimethyl- 000122-09-8 Benzenethiol 000108-98-5 Benzoic acid, 2-hydroxy-, compd.