Epigenetic Inactivation of a Cluster of Genes Flanking MLH1 in Microsatellite-Unstable Colorectal Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(APOCI, -C2, and -E and LDLR) and the Genes C3, PEPD, and GPI (Whole-Arm Translocation/Somatic Cell Hybrids/Genomic Clones/Gene Family/Atherosclerosis) A
Proc. Natl. Acad. Sci. USA Vol. 83, pp. 3929-3933, June 1986 Genetics Regional mapping of human chromosome 19: Organization of genes for plasma lipid transport (APOCI, -C2, and -E and LDLR) and the genes C3, PEPD, and GPI (whole-arm translocation/somatic cell hybrids/genomic clones/gene family/atherosclerosis) A. J. LUSIS*t, C. HEINZMANN*, R. S. SPARKES*, J. SCOTTt, T. J. KNOTTt, R. GELLER§, M. C. SPARKES*, AND T. MOHANDAS§ *Departments of Medicine and Microbiology, University of California School of Medicine, Center for the Health Sciences, Los Angeles, CA 90024; tMolecular Medicine, Medical Research Council Clinical Research Centre, Harrow, Middlesex HA1 3UJ, United Kingdom; and §Department of Pediatrics, Harbor Medical Center, Torrance, CA 90509 Communicated by Richard E. Dickerson, February 6, 1986 ABSTRACT We report the regional mapping of human from defects in the expression of the low density lipoprotein chromosome 19 genes for three apolipoproteins and a lipopro- (LDL) receptor and is strongly correlated with atheroscle- tein receptor as well as genes for three other markers. The rosis (15). Another relatively common dyslipoproteinemia, regional mapping was made possible by the use of a reciprocal type III hyperlipoproteinemia, is associated with a structural whole-arm translocation between the long arm of chromosome variation of apolipoprotein E (apoE) (16). Also, a variety of 19 and the short arm of chromosome 1. Examination of three rare apolipoprotein deficiencies result in gross perturbations separate somatic cell hybrids containing the long arm but not of plasma lipid transport; for example, apoCII deficiency the short arm of chromosome 19 indicated that the genes for results in high fasting levels oftriacylglycerol (17). -

Harnessing the Power of Epigenetics for Targeted Nutrition
SPONSOR FEATURE SPONSOR FEATURE NESTLÉ RESEARCH: Harnessing the power of epigenetics for targeted nutrition Authors Diet and genomes interact. Because of its contin- products that are nutritious, safe, promote and Martin Kussmann*, Jennifer Dean*, uous and lifelong impact, nutrition might be the maintain health, and prevent disease. With the Rondo P. Middleton†, Peter J. van most important environmental factor for human advent and development of many new technolo- Bladeren* & Johannes le Coutre* health. Molecular nutrition research strives gies, molecular nutrition research has opened to understand this interaction. Nutrigenetics up new doors of understanding – not only con- *Nestlé Research Center, addresses how an individual’s genetic make-up cerning which dietary components may impact 1000 Lausanne 26, Switzerland. leads to a predisposition for nutritional health; an organism’s health, but also how. These new †Nestlé Purina Research, St. Louis, MO, nutrigenomics (encompassing transcriptomics, insights have allowed a greater understanding 63164 USA. proteomics and metabonomics) asks how nutri- of the systems involved at multiple levels, from tion modulates gene expression; and epigenetics whole animal to the cellular and molecular lev- provides insights into a new level of regulation els. Although challenges in this area still remain, involving mechanisms of development, parental the main focus continues to be the improvement gene imprinting and metabolic programming that of health through diet. Modern molecular nutri- are beyond genetic control. tional research aims at health promotion, dis- Tailoring diets and nutrient compositions to the ease prevention, performance improvement and needs of consumer groups that share the same benefit-risk assessment1. health state, life stage or life style is a main goal Nutrition has conventionally been considered of current nutritional research. -

The Circular Genetic Map of Phage 813 by Ron Baker and Irwin Tessman
THE CIRCULAR GENETIC MAP OF PHAGE 813 BY RON BAKER AND IRWIN TESSMAN DEPARTMENT OF BIOLOGICAL SCIENCES, PURDUE UNIVERSITY, LAFAYETTE, INDIANA Communicated by S. E. Luria, July 24, 1967 Because of its small size1 phage S13 is suitable for a study of all its genes and their functions. With this aim, conditionally lethal mutants of the suppressible (su) and temperature-sensitive (t) type have been isolated and classified into seven complementation groups, and the general function of five of the genes implicitly defined by these complementation groups has been determined.8-10 This paper is concerned with the mapping of the seven known phage genes, with the ultimate aim of understanding both the organization of the phage genome and the mechanism by which it undergoes genetic recombination. Although the DNA molecule of S13, as shown for the closely related phage ,X174, is physically circular both in the single-stranded form of the mature virus11 and in the double-stranded replicative form,12' 13 this does not necessarily imply that the genetic map should also be circular since circular DNA is neither necessary nor sufficient for genetic circularity. On the one hand, a linear DNA molecule can give rise to a circular genetic map if the nucleotide sequences are circularly permuted;14 an example is the T2-T4 phage system.'5-17 On the other hand, a circular DNA can yield a linear map if recombination involves opening of the ring at a unique site, as seems to occur for phage X, which forms a closed DNA molecule after infections yet has a linear vegetative and prophage genetic map.19 In this report it will be shown that the genetic map of S13, as determined entirely by 3-factor crosses, is indeed circular, and, as might be expected for a circular genome, the occurrence of double recombination events appears to be the rule. -

Expression-Based Genetic/Physical Maps of Single-Nucleotide Polymorphisms Identified by the Cancer Genome Anatomy Project
Downloaded from genome.cshlp.org on October 11, 2021 - Published by Cold Spring Harbor Laboratory Press Resource Expression-based Genetic/Physical Maps of Single-Nucleotide Polymorphisms Identified by the Cancer Genome Anatomy Project Robert Clifford, Michael Edmonson, Ying Hu, Cu Nguyen, Titia Scherpbier, and Kenneth H. Buetow1 Laboratory of Population Genetics, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892 USA SNPs (Single-Nucleotide Polymorphisms), the most common DNA variant in humans, represent a valuable resource for the genetic analysis of cancer and other illnesses. These markers may be used in a variety of ways to investigate the genetic underpinnings of disease. In gene-based studies, the correlations between allelic variants of genes of interest and particular disease states are assessed. An extensive collection of SNP markers may enable entire molecular pathways regulating cell metabolism, growth, or differentiation to be analyzed by this approach. In addition, high-resolution genetic maps based on SNPs will greatly facilitate linkage analysis and positional cloning. The National Cancer Institute’s CGAP-GAI (Cancer Genome Anatomy Project Genetic Annotation Initiative) group has identified 10,243 SNPs by examining publicly available EST (Expressed Sequence Tag) chromatograms. More than 6800 of these polymorphisms have been placed on expression-based integrated genetic/physical maps. In addition to a set of comprehensive SNP maps, we have produced maps containing single nucleotide polymorphisms in genes expressed in breast, colon, kidney, liver, lung, or prostate tissue. The integrated maps, a SNP search engine, and a Java-based tool for viewing candidate SNPs in the context of EST assemblies can be accessed via the CGAP-GAI web site (http://cgap.nci.nih.gov/GAI/). -

HUMAN GENE MAPPING WORKSHOPS C.1973–C.1991
HUMAN GENE MAPPING WORKSHOPS c.1973–c.1991 The transcript of a Witness Seminar held by the History of Modern Biomedicine Research Group, Queen Mary University of London, on 25 March 2014 Edited by E M Jones and E M Tansey Volume 54 2015 ©The Trustee of the Wellcome Trust, London, 2015 First published by Queen Mary University of London, 2015 The History of Modern Biomedicine Research Group is funded by the Wellcome Trust, which is a registered charity, no. 210183. ISBN 978 1 91019 5031 All volumes are freely available online at www.histmodbiomed.org Please cite as: Jones E M, Tansey E M. (eds) (2015) Human Gene Mapping Workshops c.1973–c.1991. Wellcome Witnesses to Contemporary Medicine, vol. 54. London: Queen Mary University of London. CONTENTS What is a Witness Seminar? v Acknowledgements E M Tansey and E M Jones vii Illustrations and credits ix Abbreviations and ancillary guides xi Introduction Professor Peter Goodfellow xiii Transcript Edited by E M Jones and E M Tansey 1 Appendix 1 Photographs of participants at HGM1, Yale; ‘New Haven Conference 1973: First International Workshop on Human Gene Mapping’ 90 Appendix 2 Photograph of (EMBO) workshop on ‘Cell Hybridization and Somatic Cell Genetics’, 1973 96 Biographical notes 99 References 109 Index 129 Witness Seminars: Meetings and publications 141 WHAT IS A WITNESS SEMINAR? The Witness Seminar is a specialized form of oral history, where several individuals associated with a particular set of circumstances or events are invited to meet together to discuss, debate, and agree or disagree about their memories. The meeting is recorded, transcribed, and edited for publication. -

'Morbid Anatomy' of the Human Genome
Med. Hist. (2014), vol. 58(3), pp. 315–336. c The Author 2014. Published by Cambridge University Press 2014 doi:10.1017/mdh.2014.26 The ‘Morbid Anatomy’ of the Human Genome: Tracing the Observational and Representational Approaches of Postwar Genetics and Biomedicine The William Bynum Prize Essay ANDREW J. HOGAN* Department of History, Humanities Center 225, Creighton University, 2500 California Plaza, Omaha, NE 68178, USA Abstract: This paper explores evolving conceptions and depictions of the human genome among human and medical geneticists during the postwar period. Historians of science and medicine have shown significant interest in the use of informational approaches in postwar genetics, which treat the genome as an expansive digital data set composed of three billion DNA nucleotides. Since the 1950s, however, geneticists have largely interacted with the human genome at the microscopically visible level of chromosomes. Mindful of this, I examine the observational and representational approaches of postwar human and medical genetics. During the 1970s and 1980s, the genome increasingly came to be understood as, at once, a discrete part of the human anatomy and a standardised scientific object. This paper explores the role of influential medical geneticists in recasting the human genome as being a visible, tangible, and legible entity, which was highly relevant to traditional medical thinking and practice. I demonstrate how the human genome was established as an object amenable to laboratory and clinical research, and argue that the observational and representational approaches of postwar medical genetics reflect, more broadly, the interdisciplinary efforts underlying the development of contemporary biomedicine. Keywords: Medical genetics, Biomedicine, Human genome, Chromosomes, Morbid anatomy Introduction In 1982, Victor McKusick, Physician-in-Chief of the Johns Hopkins University School of Medicine, published a commentary entitled, ‘The Human Genome Through the Eyes of a Clinical Geneticist’. -

The First Genetic Linkage Map for Fraxinus Pennsylvanica and Syntenic Relationships with Four Related Species
Plant Molecular Biology (2019) 99:251–264 https://doi.org/10.1007/s11103-018-0815-9 The first genetic linkage map for Fraxinus pennsylvanica and syntenic relationships with four related species Di Wu1 · Jennifer Koch2 · Mark Coggeshall3,4 · John Carlson1 Received: 13 August 2018 / Accepted: 15 December 2018 / Published online: 2 January 2019 © Springer Nature B.V. 2019 Abstract Key message The genetic linkage map for green ash (Fraxinus pennsylvanica) contains 1201 DNA markers in 23 linkage groups spanning 2008.87cM. The green ash map shows stronger synteny with coffee than tomato. Abstract Green ash (Fraxinus pennsylvanica) is an outcrossing, diploid (2n = 46) hardwood tree species, native to North America. Native ash species in North America are being threatened by the rapid spread of the emerald ash borer (EAB, Agrilus planipennis), an invasive pest from Asia. Green ash, the most widely distributed ash species, is severely affected by EAB infestation, yet few genomic resources for genetic studies and improvement of green ash are available. In this study, a total of 5712 high quality single nucleotide polymorphisms (SNPs) were discovered using a minimum allele frequency of 1% across the entire genome through genotyping-by-sequencing. We also screened hundreds of genomic- and EST-based microsatellite markers (SSRs) from previous de novo assemblies (Staton et al., PLoS ONE 10:e0145031, 2015; Lane et al., BMC Genom 17:702, 2016). A first genetic linkage map of green ash was constructed from 90 individuals in a full-sib family, combining 2719 SNP and 84 SSR segregating markers among the parental maps. The consensus SNP and SSR map contains a total of 1201 markers in 23 linkage groups spanning 2008.87 cM, at an average inter-marker distance of 1.67 cM with a minimum logarithm of odds of 6 and maximum recombination fraction of 0.40. -

Snps) in Genes of the Alcohol Dehydrogenase, Glutathione S-Transferase, and Nicotinamide Adenine Dinucleotide, Reduced (NADH) Ubiquinone Oxidoreductase Families
J Hum Genet (2001) 46:385–407 © Jpn Soc Hum Genet and Springer-Verlag 2001 ORIGINAL ARTICLE Aritoshi Iida · Susumu Saito · Akihiro Sekine Takuya Kitamoto · Yuri Kitamura · Chihiro Mishima Saori Osawa · Kimie Kondo · Satoko Harigae Yusuke Nakamura Catalog of 434 single-nucleotide polymorphisms (SNPs) in genes of the alcohol dehydrogenase, glutathione S-transferase, and nicotinamide adenine dinucleotide, reduced (NADH) ubiquinone oxidoreductase families Received: March 15, 2001 / Accepted: April 6, 2001 Abstract An approach based on development of a large Introduction archive of single-nucleotide polymorphisms (SNPs) throughout the human genome is expected to facilitate large-scale studies to identify genes associated with drug Human genetic variations result from a dynamic process efficacy and side effects, or susceptibility to common over time that includes sudden mutations, random genetic diseases. We have already described collections of SNPs drift, and, in some cases, founder effects. Variations at a present among various genes encoding drug-metabolizing single gene locus are useful as markers of individual risk for enzymes. Here we report SNPs for such enzymes at adverse drug reactions or susceptibility to complex diseases additional loci, including 8 alcohol dehydrogenases, 12 (for reviews, see Risch and Merikangas 1996; Kruglyak glutathione S-transferases, and 18 belonging to the NADH- 1997; McCarthy and Hilfiker 2000). Common types of ubiquinone oxidoreductase family. Among DNA samples sequence variation in the human genome include single- from 48 Japanese volunteers, we identified a total of 434 nucleotide polymorphisms (SNPs), insertion/deletion SNPs at these 38 loci: 27 within coding elements, 52 in 59 polymorphisms, and variations in the number of repeats of flanking regions, five in 59 untranslated regions, 293 in certain motifs (e.g., microsatellites and variable number of introns, 20 in 39 untranslated regions, and 37 in 39 flanking tandem repeat loci). -
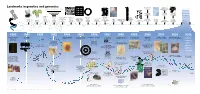
Landmarks in Genetics and Genomics
0 ++- ---+ BC PR • --- -+-t--.--+------ o,ou 1J.T33.? n.e -- -- - 4 Landmarks in genetics and genomics !!!!! U C AG +++j/GA T C US National Research U Phe Tyr Cys International Nucleotide Ser C Council issues report on U Leu stop stop A Sequence Database - stop Trp - G Mapping and Sequencing the U Consortium formed Leu Pro His Arg C Human Genome 0 5' C Gln A 3' G Human U 5' lle Asn Ser STS Markers Thr C A Met Lys Arg A G Muscular-dystrophy Development New strands Sequence-tagged Asp U of yeast artificial Genome G Val Ala Gly C gene identified sites (STS) mapping Glu A chromosome (YAC) G by positional cloning concept established cloning Archibald Garrod Project formulates Alfred Henry Oswald Avery, Colin MacLeod James Watson and Stanley Cohen and Frederick Sanger, First public Desired fragment the concept Sturtevant and Maclyn McCarty Francis Crick Marshall Nirenberg, Herbert Boyer The Belmont Report Allan Maxam discussion strands Gregor Mendel of human makes demonstrate that DNA describe the Har Gobind Khorana and develop on the use of and Walter Gilbert GenBank First human disease gene — for of sequencing First automated First-generation Cystic-fibrosis discovers Rediscovery of inborn errors the first linear is the double-helical Robert Holley determine recombinant human subjects develop DNA-sequencing database Huntington’s disease — is mapped the human The polymerase DNA-sequencing instrument human genetic Human Genome gene identified by laws of genetics Mendel’s work of metabolism map of genes hereditary material structure of DNA the genetic code DNA technology in research is issued methods established with DNA markers genome chain reaction (PCR) is invented developed map developed Organization (HUGO) formed positional cloning 1865 1900 1905 1913 1944 1953 1966 1972 1974 1977 1982 1983 1984 1985 1986 1987 1988 1989 1990 2003 +-+-o-----+---+ 1 I~I "-.■ u ''\. -

Epigenetics LSU 1/13/16
Epigenetics LSU 1/13/16 Epigenetics David Clark Professor of Pediatrics Objectives 1. …understand why identical twins are never identical. 2. …recognize environmental factors that alter gene expression. 3. …be able to incorporate “epigenetic thinking” into their daily practice Disclosure I have ~25,000 “functional” genes At least 40 have a mutation not present in either of my parents Fortunately my daughters and grandchildren did not inherit a transformed gene. I take antioxidants and fish oil in an attempt to fight environmental factors AAP Priority New Vocabulary Genomics Proteomics Pharmacogenetics Pharmacogenomics Biophotonics Epigenetics Epigenetics What is epigenetics? Why now? How does it affect the individual? Why do Physicians need to be aware? Epigenetics Vocabulary DNA methylation Evolutionary capacitance GenomePlex Histone code Nutriepigenomics Preformationism Sirtuin Synthetic genetic array Brief Timeline of Cell Biology Cell Robert Hooke 1653 RBC Nucleus Antonie van Leeuwenhook 1682 Nucleus named Robert Brown (particles) 1831 DNA Friedrich Miesher 1869 Bioblasts Richard Attman 1880 Mitochondria Carl Benda 1899 DNA composition Phoebus Levene 1919 “Epigenetics” Conrad Waddington 1942 DNA structure James Watson/Francis Crick 1953 Conrad Hal Waddington Change in Gene Expression not Associated with a change of the DNA Sequence Epi- From Greek above upon over nearby outer besides among in addition to attached to 328 English words Adrenalin --- Epinephrine Epicardium Epicanthal Epidermis Epigastric -

The First Genetic Linkage Map for Fraxinus Pennsylvanica and Syntenic Relationships with Four Related Species
bioRxiv preprint doi: https://doi.org/10.1101/365676; this version posted July 9, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The first genetic linkage map for Fraxinus pennsylvanica and syntenic relationships with four related species Authors: Di Wu1, Jennifer Koch2, Mark Coggeshall3,4, John Carlson1* 1 Department of Ecosystem Science and Management, Pennsylvania State University, University Park, PA 16802 USA 2 USDA Forest Service, Northern Research Station, Project NRS-16. 359 Main Road, Delaware, OH 43015 USA 3 Department of Forestry, Center for Agroforestry, University of Missouri, Columbia, MO 65211 USA 4 USDA Forest Service, Northern Research Station, Hardwood Tree Improvement and Regeneration Center, Project NRS-14. 715 W. State Street, West Lafayette, IN 47907 USA *Communicating Author, jec16@psu,edu Abstract Green ash (Fraxinus pennsylvanica) is an outcrossing, diploid (2n=46) hardwood tree species, native to North America. Native ash species in North America are being threatened by the rapid invasion of emerald ash borer (EAB, Agrilus planipennis) from Asia. Green ash, the most widely distributed ash species, is severely affected by EAB infestation, yet few resources for genetic studies and improvement of green ash are available. In this study, a total of 5,712 high quality single nucleotide polymorphisms (SNPs) were discovered using a minimum allele frequency of 1% across the entire genome through genotyping-by-sequencing. We also screened hundreds of genomic- and EST-based microsatellite markers (SSRs) from previous de novo assemblies (Staton et al. 2015; Lane et al. -

Single Nucleotide Polymorphism in Genome-Wide Association of Human Population: a Tool for Broad Spectrum Service
The Egyptian Journal of Medical Human Genetics (2013) 14, 123–134 Ain Shams University The Egyptian Journal of Medical Human Genetics www.ejmhg.eg.net www.sciencedirect.com REVIEW Single nucleotide polymorphism in genome-wide association of human population: A tool for broad spectrum service Mohd Fareed, Mohammad Afzal * Human Genetics and Toxicology Lab., Section of Genetics, Department of Zoology, Faculty of Life Sciences, Aligarh Muslim University, Aligarh 202002, Uttar Pradesh, India Received 2 August 2012; accepted 12 August 2012 Available online 25 September 2012 KEYWORDS Abstract Genome-wide patterns of variation across individuals provide most powerful source of SNPs; data for uncovering the history of migration, expansion, and adaptation of the human population. Genetic variation; The arrival of new technologies that type more than millions of the single nucleotide polymor- Human population; phisms (SNPs) in a single experiment has made SNP in genome-wide association (GWA) assay a Disease risk; prudent venture. SNPs represent the most widespread type of sequence variation in genomes, Drug development and known as valuable genetic markers for revealing the evolutionary history and common genetic polymorphisms that explain the heritable risk for common diseases. Characterizing the nature of gene variation in human populations and assembling an extensive catalog of SNPs in candidate genes in association with particular diseases are the major goals of human genetics. In this article we explore the recent discovery of SNP–GWA to revolutionize not only the process of genetic var- iation and disease detection but also the convention of preventative and curative medicine for future prospects. Ó 2012 Ain Shams University.