Downloaded in PDB Format from Protein Conducted Setting the Maximum Heavy Atom RMSD Data Bank (
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Shexiang Baoxin Pill, a Traditional Chinese Herbal Formula, Rescues the Cognitive Impairments in APP/ PS1 Transgenic Mice
ORIGINAL RESEARCH published: 14 July 2020 doi: 10.3389/fphar.2020.01045 Shexiang Baoxin Pill, a Traditional Chinese Herbal Formula, Rescues the Cognitive Impairments in APP/ PS1 Transgenic Mice † † Wei-Hui Hu 1,2,3 , Shing-Hung Mak 1,2 , Zhong-Yu Zheng 1,2, Ying-Jie Xia 1,2, Miranda Li Xu 1,2, Ran Duan 1,2,3, Tina Ting-Xia Dong 1,2,3, Shao-Ping Li 4, Chang-Sen Zhan 5,6, Xiao-Hui Shang 5,6 and Karl Wah-Keung Tsim 1,2,3* 1 Shenzhen Key Laboratory of Edible and Medicinal Bioresources, HKUST Shenzhen Research Institute, Shenzhen, China, 2 Division of Life Science and Center for Chinese Medicine and State Key Laboratory of Molecular Neuroscience, The Hong Kong University of Science and Technology, Hong Kong, Hong Kong, 3 Joint Laboratory of Guangdong Province and Hong Kong Region on Marine Bioresource Conservation and Exploitation, College of Marine Sciences, South China Agricultural University, Guangzhou, China, 4 Institute of Chinese Medical Sciences, University of Macau, Macau, Macau, 5 Shanghai Edited by: Engineering Research Center for Innovation of Solid Preparation of TCM, Shanghai, China, 6 Shanghai Hutchison Qiaobing Huang, Pharmaceuticals Ltd., Shanghai, China Southern Medical University, China Reviewed by: Background: Shexiang Baoxin Pill (SBP), a formulated traditional Chinese medicine Bing-Xing Pan, Nanchang University, China (TCM), has been widely used to treat cardiovascular diseases for years. This herbal Wenda Xue, mixture has been shown to promote differentiation of cultured neuronal cells. Here, we Nanjing University of Chinese aimed to investigate the effects of SBP in attenuating cognitive impairment in APP/PS1 Medicine, China *Correspondence: transgenic mice. -

Β-Phenylethylamines and the Isoquinoline Alkaloids
-Phenylethylamines and the isoquinoline alkaloids Kenneth W. Bentley Marrview, Tillybirloch, Midmar, Aberdeenshire, UK AB51 7PS Received (in Cambridge, UK) 28th November 2000 First published as an Advance Article on the web 7th February 2001 Covering: July 1999 to June 2000. Previous review: Nat. Prod. Rep., 2000, 17, 247. 1 Introduction 2 -Phenylethylamines 3 Isoquinolines 4 Naphthylisoquinolines 5 Benzylisoquinolines 6 Bisbenzylisoquinolines 7 Pavines and isopavines 8 Berberines and tetrahydoberberines 9 Protopines 10 Phthalide-isoquinolines 11 Other modified berberines 12 Emetine and related alkaloids 13 Benzophenanthridines 14 Aporphinoid alkaloids 14.1 Proaporphines 14.2 Aporphines 14.3 Aporphine–benzylisoquinoline dimers 14.4 Phenanthrenes 14.5 Oxoaporphines 14.6 Dioxoaporphines 14.7 Aristolochic acids and aristolactams 14.8 Oxoisoaporphines 15 Alkaloids of the morphine group 16 Colchicine and related alkaloids 17 Erythrina alkaloids 17.1 Erythrinanes 17.2 Cephalotaxine and related alkaloids 18 Other isoquinolines 19 References 1 Introduction Reviews of the occurrence of isoquinoline alkaloids in some plant species 1,2 and of recent developments in the chemistry and synthesis of alkaloids of these groups 3–6 have been published. 2 -Phenylethylamines β-Phenylethylamine, tyramine, N-methyltyramine, hordenine, mescaline, N-methylmescaline and N,N-dimethylmescaline 1, which is reported as an alkaloid for the first time, have been isolated from an unspecified species of Turbinocarpus 7 and N-trans-feruloyltyramine has been isolated from Cananga odorata.8 The N-oxides of the known alkaloid culantraramine 2 and the unknown culantraraminol 3, together with the related avicennamine 4 have been isolated as new alkaloids from Zanthoxylum avicennae.9 Three novel amides of dehydrotyr- leucine and proline respectively. -

In Early Alzheimer's Disease De
Protocol I8D-MC-AZFD(a) A Randomized, Double-Blind, Delayed-Start Study of LY3314814 (AZD3293) in Early Alzheimer’s Disease Dementia (Extension of Study AZES, The AMARANTH Study) NCT02972658 Approval Date: 06-Feb-2018 I8D-MC-AZFD(a) Clinical Protocol Page 1 Protocol I8D-MC-AZFD(a) A Randomized, Double-Blind, Delayed-Start Study of LY3314814 (AZD3293) in Early Alzheimer’s Disease Dementia (Extension of Study AZES, The AMARANTH Study) Confidential Information The information contained in this document is confidential and is intended for the use of clinical investigators. It is the property of Eli Lilly and Company or its subsidiaries and should not be copied by or distributed to persons not involved in the clinical investigation of LY3314814, unless such persons are bound by a confidentiality agreement with Eli Lilly and Company or its subsidiaries. Note to Regulatory Authorities: This document may contain protected personal data and/or commercially confidential information exempt from public disclosure. Eli Lilly and Company requests consultation regarding release/redaction prior to any public release. In the United States, this document is subject to Freedom of Information Act (FOIA) Exemption 4 and may not be reproduced or otherwise disseminated without the written approval of Eli Lilly and Company or its subsidiaries. LY3314814 (Lanabecestat) Study AZFD is a Phase 3 study designed to test whether LY3314814 will slow disease progression in patients with early Alzheimer’s Disease randomized in Study AZES. Eli Lilly and Company Indianapolis, Indiana USA 46285 Protocol Electronically Signed and Approved by Lilly on date provided below. Approval Date: 06-Feb-2018 GMT LY3314814 I8D-MC-AZFD(a) Clinical Protocol Page 2 Table of Contents Section Page 1. -

The “Rights” of Precision Drug Development for Alzheimer's Disease
Cummings et al. Alzheimer's Research & Therapy (2019) 11:76 https://doi.org/10.1186/s13195-019-0529-5 REVIEW Open Access The “rights” of precision drug development for Alzheimer’s disease Jeffrey Cummings1*, Howard H. Feldman2 and Philip Scheltens3 Abstract There is a high rate of failure in Alzheimer’s disease (AD) drug development with 99% of trials showing no drug- placebo difference. This low rate of success delays new treatments for patients and discourages investment in AD drug development. Studies across drug development programs in multiple disorders have identified important strategies for decreasing the risk and increasing the likelihood of success in drug development programs. These experiences provide guidance for the optimization of AD drug development. The “rights” of AD drug development include the right target, right drug, right biomarker, right participant, and right trial. The right target identifies the appropriate biologic process for an AD therapeutic intervention. The right drug must have well-understood pharmacokinetic and pharmacodynamic features, ability to penetrate the blood-brain barrier, efficacy demonstrated in animals, maximum tolerated dose established in phase I, and acceptable toxicity. The right biomarkers include participant selection biomarkers, target engagement biomarkers, biomarkers supportive of disease modification, and biomarkers for side effect monitoring. The right participant hinges on the identification of the phase of AD (preclinical, prodromal, dementia). Severity of disease and drug mechanism both have a role in defining the right participant. The right trial is a well-conducted trial with appropriate clinical and biomarker outcomes collected over an appropriate period of time, powered to detect a clinically meaningful drug-placebo difference, and anticipating variability introduced by globalization. -

Relief of Hypersensitivity After Nerve Injury from Systemic Donepezil
Saranya devi Relief of Hypersensitivity after Nerve Injury from ALN Systemic Donepezil Involves Spinal Cholinergic and γ-Aminobutyric Acid Mechanisms Spinal ACh and GABA Mechanisms for Donepezil Analgesia Masafumi Kimura, M.D.,* Ken-ichiro Hayashida, D.V.M., Ph.D.,† James C. Eisenach, M.D.,‡ KIMURA ET AL. Shigeru Saito M.D.,§ Hideaki Obata, M.D.‖ XX ABSTRACT What We Already Know about This Topic • Donepezil increases central nervous system acetylcholine lev- Downloaded from http://pubs.asahq.org/anesthesiology/article-pdf/118/1/173/259568/0000542-201301000-00030.pdf by guest on 28 September 2021 10.1097/ALN.0b013e318277a81c Background: Evoking spinal release of acetylcholine (ACh) els and is used for treatment of dementia produces antinociception in normal animals and reduces • Increases in spinal acetylcholine levels have been implicated in the relief of neuropathic pain hypersensitivity after nerve injury, and some studies suggest January that ACh-mediated analgesia relies on γ-aminobutyric acid (GABA)-ergic signaling in the spinal cord. In this study, the authors tested the spinal mechanisms underlying the anti- What This Article Tells Us That Is New 118 hypersensitivity effects of donepezil, a central nervous sys- • Donepezil increased spinal acetylcholine levels and γ- tem–penetrating cholinesterase inhibitor, in a rat model of aminobutyric acid levels to reduce nociceptive responses after nerve injury and represents a potential therapeutic pathway to neuropathic pain. reduce pain after nerve injury Methods: Male Sprague-Dawley rats were anesthetized, and L5 spinal nerve ligation was performed unilaterally. With- A antagonist; and CGP 35348 (30 μg), a γ-aminobutyric drawal threshold to a paw pressure test was measured before acid receptor type B antagonist. -

Tor Amendment Tagoor Laboratories Private Limited
ToR Amendment Tagoor Laboratories Private Limited ANNEXURE – I Details of Products as per Granted ToR and Amendment Required Production Capacity S. Ton /Month Therapeutic Product Name No As per Amendment category granted ToR Required 1 Abacavir sulfate 2.0 2.0 Anti HIV 2 Amitriptyline 5.0 10.0 Antidepressant 3 Atrovastatin Calcium 5.0 5.0 Hypercholesterolemia 4 Bupropion 5.0 5.0 Anti depressant 5 carisoprodol 2.0 2.0 Muscle Relaxant 6 Clopidogrelbisulfate 5.0 5.0 Antithrombotic 7 Cyclobenzaprine HCl 5.0 5.0 Muscle relaxant 8 Cyproheptadine HCl 5.0 10.0 Anti allergic 9 Desloratadine 5.0 5.0 Antihistamine 10 Domperidone 20.0 30.0 Anti emetic 11 Domperidone maleate 2.0 2.0 Anti emetic 12 Donepezil HCl 1.0 1.0 Alzheimer’s disease 13 Ebastine 5.0 5.0 Anti allergic 14 Esomeprazole Sodium 3.0 3.0 Anti ulcerative Esomeprazole Magnesium 3.0 3.0 Anti ulcerative 15 trihydrate 16 Fexofenadine Hydrochloride 6.0 15.0 Anti histamine 17 Haloperidol 2.0 2.0 Antipsychotic 18 Itopride Hydrochloride 2.0 2.0 Antispasmodics 19 Itraconazole 10.0 10.0 Antifungal 20 Ketrolac Tromethane 2.0 2.0 Anti Inflammatory 21 Lansoprazole 10.0 10.0 Ant ulcerative 22 Loperamide Hydrochloride 10.0 10.0 Anti diarrhea agent 23 Losartan Potassium 6.0 15.0 Anti Hypertensive 24 Nebivolol HCl 2.0 2.0 Anti Hypertensive 25 Nortriptyline HCl 2.0 2.0 Anti depressant 26 Omeprazole 40.0 40.0 Anti ulcerative 27 Omeprazole Sodium 2.0 2.0 Ant ulcerative Omeprazole Magnesium 2.0 2.0 Ant ulcerative 28 Dihydrate 29 Oxatomide 1.0 1.0 Antihistamine Pantoprazole Sodium Sesqui 10.0 20.0 Ant ulcerative 30 Hydrate 31 Pimozide 2.0 2.0 Antipsychotic 32 Pregabalin 2.0 2.0 Epileptic 33 Quetiapine Hemifumarate 2.0 2.0 Antipsychotic Prepared By Rightsource Industrial Solutions Pvt. -

Treatment of Alzheimer's Disease and Blood–Brain Barrier Drug Delivery
pharmaceuticals Review Treatment of Alzheimer’s Disease and Blood–Brain Barrier Drug Delivery William M. Pardridge Department of Medicine, University of California, Los Angeles, CA 90024, USA; [email protected] Received: 24 October 2020; Accepted: 13 November 2020; Published: 16 November 2020 Abstract: Despite the enormity of the societal and health burdens caused by Alzheimer’s disease (AD), there have been no FDA approvals for new therapeutics for AD since 2003. This profound lack of progress in treatment of AD is due to dual problems, both related to the blood–brain barrier (BBB). First, 98% of small molecule drugs do not cross the BBB, and ~100% of biologic drugs do not cross the BBB, so BBB drug delivery technology is needed in AD drug development. Second, the pharmaceutical industry has not developed BBB drug delivery technology, which would enable industry to invent new therapeutics for AD that actually penetrate into brain parenchyma from blood. In 2020, less than 1% of all AD drug development projects use a BBB drug delivery technology. The pathogenesis of AD involves chronic neuro-inflammation, the progressive deposition of insoluble amyloid-beta or tau aggregates, and neural degeneration. New drugs that both attack these multiple sites in AD, and that have been coupled with BBB drug delivery technology, can lead to new and effective treatments of this serious disorder. Keywords: blood–brain barrier; brain drug delivery; drug targeting; endothelium; Alzheimer’s disease; therapeutic antibodies; neurotrophins; TNF inhibitors 1. Introduction Alzheimer’s Disease (AD) afflicts over 50 million people world-wide, and this health burden costs over 1% of global GDP [1]. -

Current Research Efforts in the Prevention and Treatment of Alzheimer’S Disease (AD) and Related Dementias
Current Research efforts in the prevention and treatment of Alzheimer’s Disease (AD) and Related Dementias What’s new? 1. New and More effective Biomarkers 2. New Diagnostic Framework : • New “A/T/N” Framework • New Framework conceptualizes AD as a continuum from pathophysiological, biomarker and clinical perspectives. 3. LATE (Limbic-predominant, Age-related, TDP-43 Encephalopathy) looks like Alzheimer’s disease 4. Alzheimer’s pathophysiology starts decades prior to clinical disease! 1. New and More Effective Biomarkers AD Pathology • Alzheimer’s Disease consists of two main features: • Senile plaques (β-amyloid)- earliest pathological event! • Neurofibrillary tangles (hyperphosphorylated tau)- primary culprit in cells death • Mechanism: • β-amyloid hyperphosphorylated tau tau spreads throughout the brain from neuron to neuron (Pooler et al, 2013). Cell Death Biomarkers Budson & Solomon, Practical Neurology 2012;12:88–96 PET Amyloid Imaging was approved since 2012 • Use when knowing that AD pathology is present in symptomatic patient would change management. • May detect amyloid plaques in asymptomatic patients who may not develop disease for 10-15+ years • Not paid for by Medicare or other insurance companies • Can obtain through Veterans Affairs hospitals, clinical trials/research studies, and self-pay. • Will have broader use when disease modifying therapies are available. Alzheimer’s Disease Non-AD dementia 65 year old MoCA 21 From Budson & Solomon, 2016 PET tau imaging : FDA approved May 2020 • Use when knowing that AD pathology is present in symptomatic patient would change management. • Will detect AD tau tangles in symptomatic patients and should correlate with symptoms • May detect other types of tau tangles in other dementias (not yet clear) • Not paid for by Medicare or other insurance companies • Can obtain through Veterans Affairs hospitals, clinical trials/research studies, and self-pay. -

Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration
Review Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration Rahat Ullah 1,†, Mehtab Khan 1,†, Shahid Ali Shah 1,2, Kamran Saeed 1 and Myeong Ok Kim 1,* 1 Division of Applied Life Science (BK 21), College of Natural Sciences, Gyeongsang National University, Jinju 52828, Korea; [email protected] (R.U.); [email protected] (M.K.); [email protected] (S.A.S.); [email protected] (K.S.) 2 Department of Chemistry, Sarhad University of Science & Information Technology (SUIT), Peshawar Khyber Pakhtunkhwa 25000, Pakistan * Correspondence: [email protected]; Tel.: +82-55-772-1345; Fax: +82-55-772-2656 † These authors contributed equally to this paper. Received: 7 May 2019; Accepted: 24 May 2019; Published: 28 May 2019 Abstract: All over the world, metabolic syndrome constitutes severe health problems. Multiple factors have been reported in the pathogenesis of metabolic syndrome. Metabolic disorders result in reactive oxygen species (ROS) induced oxidative stress, playing a vital role in the development and pathogenesis of major health issues, including neurological disorders Alzheimer’s disease (AD) Parkinson's disease (PD). Considerable increasing evidence indicates the substantial contribution of ROS-induced oxidative stress in neurodegenerative diseases. An imbalanced metabolism results in a defective antioxidant defense system, free radicals causing inflammation, cellular apoptosis, and tissue damage. Due to the annual increase in financial and social burdens, in addition to the adverse effects associated with available synthetic agents, treatment diversion from synthetic to natural approaches has occurred. Antioxidants are now being considered as convincing therapeutic agents against various neurodegenerative disorders. -
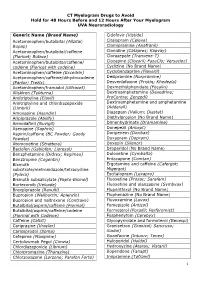
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology Generic Name (Brand Name) Cidofovir (Vistide) Acetaminophen/butalbital (Allzital; Citalopram (Celexa) Bupap) Clomipramine (Anafranil) Acetaminophen/butalbital/caffeine Clonidine (Catapres; Kapvay) (Fioricet; Butace) Clorazepate (Tranxene-T) Acetaminophen/butalbital/caffeine/ Clozapine (Clozaril; FazaClo; Versacloz) codeine (Fioricet with codeine) Cyclizine (No Brand Name) Acetaminophen/caffeine (Excedrin) Cyclobenzaprine (Flexeril) Acetaminophen/caffeine/dihydrocodeine Desipramine (Norpramine) (Panlor; Trezix) Desvenlafaxine (Pristiq; Khedezla) Acetaminophen/tramadol (Ultracet) Dexmethylphenidate (Focalin) Aliskiren (Tekturna) Dextroamphetamine (Dexedrine; Amitriptyline (Elavil) ProCentra; Zenzedi) Amitriptyline and chlordiazepoxide Dextroamphetamine and amphetamine (Limbril) (Adderall) Amoxapine (Asendin) Diazepam (Valium; Diastat) Aripiprazole (Abilify) Diethylpropion (No Brand Name) Armodafinil (Nuvigil) Dimenhydrinate (Dramamine) Asenapine (Saphris) Donepezil (Aricept) Aspirin/caffeine (BC Powder; Goody Doripenem (Doribax) Powder) Doxapram (Dopram) Atomoxetine (Strattera) Doxepin (Silenor) Baclofen (Gablofen; Lioresal) Droperidol (No Brand Name) Benzphetamine (Didrex; Regimex) Duloxetine (Cymbalta) Benztropine (Cogentin) Entacapone (Comtan) Bismuth Ergotamine and caffeine (Cafergot; subcitrate/metronidazole/tetracycline Migergot) (Pylera) Escitalopram (Lexapro) Bismuth subsalicylate (Pepto-Bismol) Fluoxetine (Prozac; Sarafem) -

Drug Candidates in Clinical Trials for Alzheimer's Disease
Hung and Fu Journal of Biomedical Science (2017) 24:47 DOI 10.1186/s12929-017-0355-7 REVIEW Open Access Drug candidates in clinical trials for Alzheimer’s disease Shih-Ya Hung1,2 and Wen-Mei Fu3* Abstract Alzheimer’s disease (AD) is a major form of senile dementia, characterized by progressive memory and neuronal loss combined with cognitive impairment. AD is the most common neurodegenerative disease worldwide, affecting one-fifth of those aged over 85 years. Recent therapeutic approaches have been strongly influenced by five neuropathological hallmarks of AD: acetylcholine deficiency, glutamate excitotoxicity, extracellular deposition of amyloid-β (Aβ plague), formation of intraneuronal neurofibrillary tangles (NTFs), and neuroinflammation. The lowered concentrations of acetylcholine (ACh) in AD result in a progressive and significant loss of cognitive and behavioral function. Current AD medications, memantine and acetylcholinesterase inhibitors (AChEIs) alleviate some of these symptoms by enhancing cholinergic signaling, but they are not curative. Since 2003, no new drugs have been approved for the treatment of AD. This article focuses on the current research in clinical trials targeting the neuropathological findings of AD including acetylcholine response, glutamate transmission, Aβ clearance, tau protein deposits, and neuroinflammation. These investigations include acetylcholinesterase inhibitors, agonists and antagonists of neurotransmitter receptors, β-secretase (BACE) or γ-secretase inhibitors, vaccines or antibodies targeting Aβ clearance or tau protein, as well as anti-inflammation compounds. Ongoing Phase III clinical trials via passive immunotherapy against Aβ peptides (crenezumab, gantenerumab, and aducanumab) seem to be promising. Using small molecules blocking 5-HT6 serotonin receptor (intepirdine), inhibiting BACE activity (E2609, AZD3293, and verubecestat), or reducing tau aggregation (TRx0237) are also currently in Phase III clinical trials. -

Retrospective Observation of Liver Function Parameters for 101 Patients Using Herbal Drugs for One Month
31 2 (2010 3 ) J Korean Oriental Med 2010;31(2):149-157 Original article 김동민1, 김회권2, 조성연3, 김용석1, 남상수1 1경희대학교 한의과대학 침구학교실, 2가평군 보건소, 3경희대학교 강남경희한방병원 침구과 Retrospective Observation of Liver Function Parameters for 101 Patients Using Herbal Drugs for One Month Dong-Min Kim 1, Hyee-Kwon Kim 2, Seong-Yeun Cho 3, Yong-Suk Kim 1, Sang-Soo Nam 1 1Dept. of Acupuncture & Moxibustion, College of Oriental Medicine, Kyung Hee University 2Gapyeong Public Health Center 3Dept. of Acupuncture & Moxibustion, Kangnam KyungHee Korean Hospital, Kyung Hee University Objective: The purpose of this study was to investigate the level of safety on liver functions when Korean herbal medicine was taken internally. Method: 101 inpatients who took Korean herbal medicine were enrolled and liver function test (aspartic aminotransferase, alanine aminotransferase, alkaline phosphatase, total bilirubin, gamma glutamyltranspeptidase, albumin, lactate dehydrogenase) was performed on admission and 1 month later. Results: In 101 patients, alkaline phosphatase and gamma glutamyltranspeptidase decreased significantly compared with the value taken on admission (p<0.05) but aspartic aminotransferase, alanine aminotransferase, total bilirubin, albumin, and lactate dehydrogenase were not significantly changed (p>0.05). In the patients who took Scutellaria baicalensis (n=34), alkaline phosphatase decreased and albumin increased significantly (p<0.05). Among the patients who took Atractylodes macrocephala (n=29), alkaline phosphatase decreased significantly (p<0.05). In the patients who took Glycyrrhiza uralensis or Paeonia lactiflora , liver function parameters were not significantly changed (p>0.05). On admission 11 patients had abnormal liver function and 2 patients had liver injury while 7 patients had abnormal liver function and 2 patients showed liver injury 1 month later.