Exome Sequencing of Patients with Histiocytoid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aplicación De La Biología De Sistemas Al Estudio De La Malaria Y Búsqueda De Biomarcadores Y Dianas Terapéuticas
Aplicación de la biología de sistemas al estudio de la malaria y búsqueda de biomarcadores y dianas terapéuticas Autora: Mireia Ferrer Almirall Máster en Bioinformática y Bioestadística Area 1-Bioinformática farmacéutica Tutores: Melchor Sánchez Martínez y Alex Sánchez Pla Profesor responsable de la asignatura: Carles Ventura Royo 02/01/2019 Esta obra está sujeta a una licencia de Reconocimiento-NoComercial- SinObraDerivada 3.0 España de Creative Commons FICHA DEL TRABAJO FINAL Aplicación de la biología de sistemas al Título del trabajo: estudio de la malaria y búsqueda de biomarcadores y dianas terapéuticas Nombre del autor: Mireia Ferrer Almirall Melchor Sánchez Martínez y Nombre del consultor/a: Alex Sánchez Pla Nombre del PRA: Carles Ventura Royo Fecha de entrega (mm/aaaa): 01/2019 Titulación: Máster en Bioinformática y Bioestadística Área del Trabajo Final: 1-Bioinformática farmacéutica Idioma del trabajo: castellano Malaria, Biología-de-sistemas, Palabras clave dianas-terapéuticas Resumen del Trabajo (máximo 250 palabras): Con la finalidad, contexto de aplicación, metodología, resultados i conclusiones del trabajo. La finalidad de este trabajo es aplicar herramientas de biología de sistemas para investigar los mecanismos implicados en la infección por el parásito de la malaria e identificar posibles biomarcadores y dianas terapéuticas. Se ha partido de una serie temporal de datos de microarrays del bazo de ratones infectados con dos cepas del parásito (NL y L) para determinar los genes que se encuentran diferencialmente expresados (DEG) respecto a ratones control. A partir de las listas de DEG obtenidas, se han utilizado herramientas de biología de sistemas en combinación con análisis de significación biológica para obtener una visión integrada de los procesos biológicos que se encuentran alterados en la enfermedad e identificar posibles biomarcadores/dianas terapéuticas. -

Low Abundance of the Matrix Arm of Complex I in Mitochondria Predicts Longevity in Mice
ARTICLE Received 24 Jan 2014 | Accepted 9 Apr 2014 | Published 12 May 2014 DOI: 10.1038/ncomms4837 OPEN Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice Satomi Miwa1, Howsun Jow2, Karen Baty3, Amy Johnson1, Rafal Czapiewski1, Gabriele Saretzki1, Achim Treumann3 & Thomas von Zglinicki1 Mitochondrial function is an important determinant of the ageing process; however, the mitochondrial properties that enable longevity are not well understood. Here we show that optimal assembly of mitochondrial complex I predicts longevity in mice. Using an unbiased high-coverage high-confidence approach, we demonstrate that electron transport chain proteins, especially the matrix arm subunits of complex I, are decreased in young long-living mice, which is associated with improved complex I assembly, higher complex I-linked state 3 oxygen consumption rates and decreased superoxide production, whereas the opposite is seen in old mice. Disruption of complex I assembly reduces oxidative metabolism with concomitant increase in mitochondrial superoxide production. This is rescued by knockdown of the mitochondrial chaperone, prohibitin. Disrupted complex I assembly causes premature senescence in primary cells. We propose that lower abundance of free catalytic complex I components supports complex I assembly, efficacy of substrate utilization and minimal ROS production, enabling enhanced longevity. 1 Institute for Ageing and Health, Newcastle University, Newcastle upon Tyne NE4 5PL, UK. 2 Centre for Integrated Systems Biology of Ageing and Nutrition, Newcastle University, Newcastle upon Tyne NE4 5PL, UK. 3 Newcastle University Protein and Proteome Analysis, Devonshire Building, Devonshire Terrace, Newcastle upon Tyne NE1 7RU, UK. Correspondence and requests for materials should be addressed to T.v.Z. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -
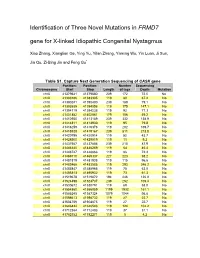
Identification of Three Novel Mutations in FRMD7 Gene for X-Linked Idiopathic Congenital Nystagmus
Identification of Three Novel Mutations in FRMD7 gene for X-linked Idiopathic Congenital Nystagmus Xiao Zhang, Xianglian Ge, Ying Yu, Yilan Zhang, Yaming Wu, Yin Luan, Ji Sun, Jia Qu, Zi-Bing Jin and Feng Gu* Table S1. Capture Next Generation Sequencing of CASK gene Position: Position: Number Sequencing Chromosome Start Stop Length of tags Depth Mutation chrX 41379641 41379880 239 172 72.0 No chrX 41383186 41383305 119 80 67.2 No chrX 41390241 41390480 239 189 79.1 No chrX 41393939 41394058 119 175 147.1 No chrX 41394119 41394238 119 92 77.3 No chrX 41401882 41402061 179 106 59.2 No chrX 41412950 41413189 239 332 138.9 No chrX 41414811 41414930 119 95 79.8 No chrX 41416259 41416378 119 202 169.7 No chrX 41418928 41419167 239 511 213.8 No chrX 41420795 41420914 119 52 43.7 No chrX 41428900 41429019 119 11 9.2 No chrX 41437567 41437806 239 210 87.9 No chrX 41446140 41446259 119 54 45.4 No chrX 41448747 41448866 119 86 72.3 No chrX 41469110 41469337 227 223 98.2 No chrX 41481819 41481938 119 115 96.6 No chrX 41483466 41483585 119 293 246.2 No chrX 41485847 41485966 119 75 63.0 No chrX 41495813 41495932 119 73 61.3 No chrX 41519678 41519872 194 246 126.8 No chrX 41524498 41524737 239 252 105.4 No chrX 41530672 41530791 119 69 58.0 No chrX 41554860 41556059 1199 1932 161.1 No chrX 41586245 41587324 1079 1044 96.8 No chrX 41598613 41598732 119 27 22.7 No chrX 41604756 41604875 119 27 22.7 No chrX 41646424 41646543 119 124 104.2 No chrX 41712364 41712483 119 37 31.1 No chrX 41782152 41782271 119 5 4.2 No Table S2. -

THE FUNCTIONAL SIGNIFICANCE of MITOCHONDRIAL SUPERCOMPLEXES in C. ELEGANS by WICHIT SUTHAMMARAK Submitted in Partial Fulfillment
THE FUNCTIONAL SIGNIFICANCE OF MITOCHONDRIAL SUPERCOMPLEXES in C. ELEGANS by WICHIT SUTHAMMARAK Submitted in partial fulfillment of the requirements For the degree of Doctor of Philosophy Dissertation Advisor: Drs. Margaret M. Sedensky & Philip G. Morgan Department of Genetics CASE WESTERN RESERVE UNIVERSITY January, 2011 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of _____________________________________________________ candidate for the ______________________degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. Dedicated to my family, my teachers and all of my beloved ones for their love and support ii ACKNOWLEDGEMENTS My advanced academic journey began 5 years ago on the opposite side of the world. I traveled to the United States from Thailand in search of a better understanding of science so that one day I can return to my homeland and apply the knowledge and experience I have gained to improve the lives of those affected by sickness and disease yet unanswered by science. Ultimately, I hoped to make the academic transition into the scholarly community by proving myself through scientific research and understanding so that I can make a meaningful contribution to both the scientific and medical communities. The following dissertation would not have been possible without the help, support, and guidance of a lot of people both near and far. I wish to thank all who have aided me in one way or another on this long yet rewarding journey. My sincerest thanks and appreciation goes to my advisors Philip Morgan and Margaret Sedensky. -

Analyzing the Function of TRAP1 in Models of Parkinson's Disease
Analyzing the function of TRAP1 in models of Parkinson’s disease Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der RWTH Aachen University zur Erlangung des akademischen Grades einer Doktorin der Naturwissenschaften genehmigte Dissertation vorgelegt von Li Zhang aus Changchun, Jilin (China) Berichter: Universitätsprofessor Dr. med. Jörg B. Schulz Universitätsprofessor Dr. rer. nat. Marc Spehr Tag der mündlichen Prüfung: 29.01.2016 Diese Dissertation ist auf den Internetseiten der Universitätsbibliothek online verfügbar. Eidesstattliche Versicherung ___________________________Zhang, Li ___________________________ Name, Vorname Matrikelnummer (freiwillige Angabe) Ich versichere hiermit an Eides Statt, dass ich die vorliegende Arbeit/Bachelorarbeit/ Masterarbeit* mit dem Titel __________________________________________________________________________Analyzing the function of TRAP1 in models of Parkinson’s disease __________________________________________________________________________Uebersetzung: Analyse der TRAP1-Funktion in Modellen fuer Morbus Parkinson __________________________________________________________________________ selbständig und ohne unzulässige fremde Hilfe erbracht habe. Ich habe keine anderen als die angegebenen Quellen und Hilfsmittel benutzt. Für den Fall, dass die Arbeit zusätzlich auf einem Datenträger eingereicht wird, erkläre ich, dass die schriftliche und die elektronische Form vollständig übereinstimmen. Die Arbeit hat in gleicher oder ähnlicher Form noch keiner Prüfungsbehörde vorgelegen. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Electron Transport Chain Activity Is a Predictor and Target for Venetoclax Sensitivity in Multiple Myeloma
ARTICLE https://doi.org/10.1038/s41467-020-15051-z OPEN Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma Richa Bajpai1,7, Aditi Sharma 1,7, Abhinav Achreja2,3, Claudia L. Edgar1, Changyong Wei1, Arusha A. Siddiqa1, Vikas A. Gupta1, Shannon M. Matulis1, Samuel K. McBrayer 4, Anjali Mittal3,5, Manali Rupji 6, Benjamin G. Barwick 1, Sagar Lonial1, Ajay K. Nooka 1, Lawrence H. Boise 1, Deepak Nagrath2,3,5 & ✉ Mala Shanmugam 1 1234567890():,; The BCL-2 antagonist venetoclax is highly effective in multiple myeloma (MM) patients exhibiting the 11;14 translocation, the mechanistic basis of which is unknown. In evaluating cellular energetics and metabolism of t(11;14) and non-t(11;14) MM, we determine that venetoclax-sensitive myeloma has reduced mitochondrial respiration. Consistent with this, low electron transport chain (ETC) Complex I and Complex II activities correlate with venetoclax sensitivity. Inhibition of Complex I, using IACS-010759, an orally bioavailable Complex I inhibitor in clinical trials, as well as succinate ubiquinone reductase (SQR) activity of Complex II, using thenoyltrifluoroacetone (TTFA) or introduction of SDHC R72C mutant, independently sensitize resistant MM to venetoclax. We demonstrate that ETC inhibition increases BCL-2 dependence and the ‘primed’ state via the ATF4-BIM/NOXA axis. Further, SQR activity correlates with venetoclax sensitivity in patient samples irrespective of t(11;14) status. Use of SQR activity in a functional-biomarker informed manner may better select for MM patients responsive to venetoclax therapy. 1 Department of Hematology and Medical Oncology, Winship Cancer Institute, School of Medicine, Emory University, Atlanta, GA, USA. -

Transcriptomic and Proteomic Landscape of Mitochondrial
TOOLS AND RESOURCES Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals Inge Ku¨ hl1,2†*, Maria Miranda1†, Ilian Atanassov3, Irina Kuznetsova4,5, Yvonne Hinze3, Arnaud Mourier6, Aleksandra Filipovska4,5, Nils-Go¨ ran Larsson1,7* 1Department of Mitochondrial Biology, Max Planck Institute for Biology of Ageing, Cologne, Germany; 2Department of Cell Biology, Institute of Integrative Biology of the Cell (I2BC) UMR9198, CEA, CNRS, Univ. Paris-Sud, Universite´ Paris-Saclay, Gif- sur-Yvette, France; 3Proteomics Core Facility, Max Planck Institute for Biology of Ageing, Cologne, Germany; 4Harry Perkins Institute of Medical Research, The University of Western Australia, Nedlands, Australia; 5School of Molecular Sciences, The University of Western Australia, Crawley, Australia; 6The Centre National de la Recherche Scientifique, Institut de Biochimie et Ge´ne´tique Cellulaires, Universite´ de Bordeaux, Bordeaux, France; 7Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden Abstract Dysfunction of the oxidative phosphorylation (OXPHOS) system is a major cause of human disease and the cellular consequences are highly complex. Here, we present comparative *For correspondence: analyses of mitochondrial proteomes, cellular transcriptomes and targeted metabolomics of five [email protected] knockout mouse strains deficient in essential factors required for mitochondrial DNA gene (IKu¨ ); expression, leading to OXPHOS dysfunction. Moreover, -

Mini-Review Phosphorylation of OXPHOS Machinery Subunits: Functional Implications in Cell Biology and Disease
YALE JOURNAL OF BIOLOGY AND MEDICINE 92 (2019), pp.523-531. Mini-Review Phosphorylation of OXPHOS Machinery Subunits: Functional Implications in Cell Biology and Disease Ernesto Castellanosa and Nathan James Lanningb,* aDepartment of Biochemistry, California State University, Los Angeles, CA; bDepartment of Biological Sciences, California State University, Los Angeles, CA The complexes of the electron transport chain and ATP synthase comprise the oxidative phosphorylation (OXPHOS†) system. The reactions of OXPHOS generate the mitochondrial membrane potential, drive the majority of ATP production in respiring cells, and contribute significantly to cellular reactive oxygen species (ROS). Regulation of OXPHOS is therefore critical to maintain cellular homeostasis. OXPHOS machinery subunits have been found to be highly phosphorylated, implicating this post-translational modification as a means whereby OXPHOS is regulated. Multiple lines of evidence now reveal the diverse mechanisms by which phosphorylation of OXPHOS machinery serve to regulate individual complex stability and activity as well as broader cellular functions. From these mechanistic studies of OXPHOS machinery phosphorylation, it is now clear that many aspects of human health and disease are potentially impacted by phosphorylation of OXPHOS complexes. This mini-review summarizes recent studies that provide robust mechanistic detail related to OXPHOS subunit phosphorylation. INTRODUCTION dysregulated bioenergetics play an important role in the etiology of many diseases, including diabetes, obesity, Maintenance of bioenergetic homeostasis is a cardiovascular disease, aging and neurodegenerative principle function of cellular metabolism as sufficient diseases such as Alzheimer’s and Parkinson’s disease energy levels must be maintained for cells to thrive [1]. [1-4]. Additionally, several mitochondrial diseases di- The complexes of the electron transport chain and ATP rectly result from deficient OXPHOS-dependent energy synthase comprise the OXPHOS system, through which production [5-7]. -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like