The Abcs of Hypertonia Management – ITB, SDR, DBS
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Management and Investigation of Neonatal Encephalopathy: 2017 Update Kathryn Martinello,1 Anthony R Hart,2 Sufin Yap,3 Subhabrata Mitra,1 Nicola J Robertson1
Review Arch Dis Child Fetal Neonatal Ed: first published as 10.1136/archdischild-2015-309639 on 6 April 2017. Downloaded from Management and investigation of neonatal encephalopathy: 2017 update Kathryn Martinello,1 Anthony R Hart,2 Sufin Yap,3 Subhabrata Mitra,1 Nicola J Robertson1 1Department of Neonatology, ABSTRACT definite aetiological diagnosis is known, and Institute for Women’s Health, This review discusses an approach to determining the hypoxic-ischaemic encephalopathy (HIE) where University College London, UK 2 cause of neonatal encephalopathy, as well as current clear diagnosis of hypoxia-ischaemia is known to Department of Neonatal and ’ Paediatric Neurology, Sheffield evidence on resuscitation and subsequent management have led to the neonate s clinical state. Children’s Hospital NHS of hypoxic-ischaemic encephalopathy (HIE). Foundation Trust, Sheffield, UK Encephalopathy in neonates can be due to varied 3 DETERMINING THE AETIOLOGY OF NE Department of Inherited aetiologies in addition to hypoxic-ischaemia. A Metabolic Diseases, Sheffield The initial stages of managing NE will be the same Children’s Hospital NHS combination of careful history, examination and the for most babies, with good resuscitation and sup- Foundation Trust, Sheffield, UK judicious use of investigations can help determine the portive management. However, as the picture cause. Over the last 7 years, infants with moderate to evolves and investigations return, clinicians should fi Correspondence to severe HIE have bene ted from the introduction of consider the aetiology of NE as this could lead to Professor Nicola J Robertson, routine therapeutic hypothermia; the number needed to specific treatments, aid with prognosis and recur- Institute for Women’s Health, treat for an additional beneficial outcome is 7 (95% CI University College London, 74 rence risk counselling, and assist with the evalu- Huntley Street, London WC1E 5 to 10). -

Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H
Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H. Long, MD, PhD,a,b Jennifer G. Fiore, MD,a,b Riaz Gillani, MD,a,b Laurie M. Douglass, MD,c Alan M. Fujii, MD,d Jodi D. Hoffman, MDe A 2-day old term male infant was found to be hypotonic and minimally abstract reactive during routine nursing care in the newborn nursery. At 40 hours of life, he was hypoglycemic and had intermittent desaturations to 70%. His mother had an unremarkable pregnancy and spontaneous vaginal delivery. The mother’s prenatal serology results were negative for infectious risk factors. Apgar scores were 9 at 1 and 5 minutes of life. On day 1 of life, he fed, stooled, and voided well. Our expert panel discusses the differential diagnosis of hypotonia in a neonate, offers diagnostic and management recommendations, and discusses the final diagnosis. DRS LONG, FIORE, AND GILLANI, birth weight was 3.4 kg (56th PEDIATRIC RESIDENTS percentile), length was 52 cm (87th aDepartment of Medicine, Boston Children’s Hospital, d e percentile), and head circumference Boston, Massachusetts; and Neonatology Section, Medical A 2-day old male infant born at Genetics Section, cDivision of Child Neurology, and 38 weeks and 4 days was found to be was 33 cm (12th percentile). His bDepartment of Pediatrics, Boston Medical Center, Boston, limp and minimally reactive during physical examination at birth was Massachusetts routine care in the newborn nursery. normal for gestational age, with Drs Long, Fiore, and Gillani conceptualized, drafted, Just 5 hours before, he had an appropriate neurologic, cardiac, and and edited the manuscript; Drs Douglass, Fujii, and appropriate neurologic status when respiratory components. -

Hereditary Spastic Paraparesis: a Review of New Developments
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.69.2.150 on 1 August 2000. Downloaded from 150 J Neurol Neurosurg Psychiatry 2000;69:150–160 REVIEW Hereditary spastic paraparesis: a review of new developments CJ McDermott, K White, K Bushby, PJ Shaw Hereditary spastic paraparesis (HSP) or the reditary spastic paraparesis will no doubt Strümpell-Lorrain syndrome is the name given provide a more useful and relevant classifi- to a heterogeneous group of inherited disorders cation. in which the main clinical feature is progressive lower limb spasticity. Before the advent of Epidemiology molecular genetic studies into these disorders, The prevalence of HSP varies in diVerent several classifications had been proposed, studies. Such variation is probably due to a based on the mode of inheritance, the age of combination of diVering diagnostic criteria, onset of symptoms, and the presence or other- variable epidemiological methodology, and wise of additional clinical features. Families geographical factors. Some studies in which with autosomal dominant, autosomal recessive, similar criteria and methods were employed and X-linked inheritance have been described. found the prevalance of HSP/100 000 to be 2.7 in Molise Italy, 4.3 in Valle d’Aosta Italy, and 10–12 Historical aspects 2.0 in Portugal. These studies employed the In 1880 Strümpell published what is consid- diagnostic criteria suggested by Harding and ered to be the first clear description of HSP.He utilised all health institutions and various reported a family in which two brothers were health care professionals in ascertaining cases aVected by spastic paraplegia. The father was from the specific region. -

Effectiveness of Myofascial Release on Spasticity and Lower Extremity
hysical M f P ed l o ic a in n r e u & o R J International Journal of Kumar and Vaidya, Int J Phys Med Rehabil 2014, 3:1 l e a h n a DOI: 10.4172/2329-9096.1000253 o b i t i l a ISSN: 2329-9096i t a n r t i e o t n n I Physical Medicine & Rehabilitation Research Article Open Access Effectiveness of Myofascial Release on Spasticity and Lower Extremity Function in Diplegic Cerebral Palsy: Randomized Controlled Trial Chandan Kumar1* and Snehashri N Vaidya2 1Associate Professor, Smt Kashibai Navale College of Physiotherapy, Narhe, Pune, Maharashtra, India 2MGM’S School of Physiotherapy, Aurangabad, India Abstract Purpose: To find out the effectiveness of Myofascial Release in combination with conventional physiotherapy on spasticity of calf, hamstring and adductors of hip and on lower extremity function in spastic diplegic subjects. Methodology: 30 spastic diplegic subjects of age group 2-8 years were taken by random sampling method from MGM College and other private clinics in Aurangabad. 15 subjects were assigned in each group. Group A: Myofascial release and conventional PT treatment. Group B: conventional PT treatment. Both the groups received training for 4 weeks. Baseline and Post treatment measures of Modified Ashworth Scale (MAS), Modified Tardieu Scale (MTS) and Gross Motor Function Test (GMFM-88) were evaluated. Results: Mean difference of MAS and R2 value of MTS in group A was more than group B, for calf, hamstring and adductors, whereas GMFM showed nearly equal improvement in both groups. Conclusion: Overall, it can be concluded from our study that the MFR along with conventional treatment reduces spasticity in calf, hamstring and adductors of hip in spastic diplegic subjects. -

Hypotonia Surestep Product Catalog Page 29 in Step with Pediatric Hypotonia
SPECIAL EDUCATIONAL SERIES DIAGNOSTIC INSIGHTS ANALYZING GAIT CHANGES GROSS MOTOR SKILLS ORTHOTIC MANAGEMENT CLI N I CAL CASE STUDIES Sponsored by an educational grant from: In Step With Pediatric Hypotonia SureStep Product Catalog Page 29 In Step With Pediatric Hypotonia Contents VIEWPOINT FROM THE EDITOR: An Unexpected Path, Mobility and More an Invaluable Perspective At the most basic level, mobility is about get- PAGE 3 ting from point A to point B. But, for many children with hypotonia, it’s about so much 4 more. FEATURES It’s about independence. It’s about con- fidence. It’s about maintaining strength, fit- ness, and healthy bones. It’s about not being Understanding Hypotonia excluded from activities enjoyed by their PAGE 4 typically developing peers. And improved mobility may have even Gait: The Cornerstone more benefits in those children whose hy- potonia is associated with social and behav- of Intervention ioral developmental delays. New research PAGE 8 has identified an association between motor skills and sociobehavioral milestones in chil- 8 The Importance of Gross dren with autism spectrum disorder, who often present with hypotonia (see “The Im- Motor Skills portance of Gross Motor Skills,” page 12). PAGE 12 This suggests that early intervention to improve gross motor skills—including or- thotic devices and physical therapy—may Orthotic Solutions for also help certain children interact more Children with Hypotonia comfortably with others. That won’t come as PAGE 16 a surprise to the clinicians and parents who 12 have personally seen it happen. This special issue is filled with evidence- Orthotic Success Stories: based information and personal success sto- Four Cases in a Series ries illustrating how effective interventions can enhance mobility in children with hy- PAGE 20 potonia. -

Cerebral Palsy
Cerebral Palsy Cerebral palsy encompasses a group of non-progressive and non-contagious motor conditions that cause physical disability in various facets of body movement. Cerebral palsy is one of the most common crippling conditions of childhood, dating to events and brain injury before, during or soon after birth. Cerebral palsy is a debilitating condition in which the developing brain is irreversibly damaged, resulting in loss of motor function and sometimes also cognitive function. Despite the large increase in medical intervention during pregnancy and childbirth, the incidence of cerebral palsy has remained relatively stable for the last 60 years. In Australia, a baby is born with cerebral palsy about every 15 hours, equivalent to 1 in 400 births. Presently, there is no cure for cerebral palsy. Classification Cerebral palsy is divided into four major classifications to describe different movement impairments. Movements can be uncontrolled or unpredictable, muscles can be stiff or tight and in some cases people have shaky movements or tremors. These classifications also reflect the areas of the brain that are damaged. The four major classifications are: spastic, ataxic, athetoid/dyskinetic and mixed. In most cases of cerebral palsy, the exact cause is unknown. Suggested possible causes include developmental abnormalities of the brain, brain injury to the fetus caused by low oxygen levels (asphyxia) or poor circulation, preterm birth, infection, and trauma. Spastic cerebral palsy leads to increased muscle tone and inability for muscles to relax (hypertonic). The brain injury usually stems from upper motor neuron in the brain. Spastic cerebral palsy is classified depending on the region of the body affected; these include: spastic hemiplegia; one side being affected, spastic monoplegia; a single limb being affected, spastic triplegia; three limbs being affected, spastic quadriplegia; all four limbs more or less equally affected. -

Treatment of Spastic Foot Deformities
TREATMENT OF SPASTIC FOOT DEFORMITIES penn neuro-orthopaedics service Table of Contents OVERVIEW Severe loss of movement is often the result of neurological disorders, Overview .............................................................. 1 such as stroke or brain injury. As a result, ordinary daily activities Treatment ............................................................. 2 such as walking, eating and dressing can be difficult and sometimes impossible to accomplish. Procedures ........................................................... 4 The Penn Neuro-Orthopaedics Service assists patients with Achilles Tendon Lengthening .........................................4 orthopaedic problems caused by certain neurologic disorders. Our Toe Flexor Releases .....................................................5 team successfully treats a wide range of problems affecting the limbs including foot deformities and walking problems due to abnormal Toe Flexor Transfer .......................................................6 postures of the foot. Split Anterior Tibialis Tendon Transfer (SPLATT) ...............7 This booklet focuses on the treatment of spastic foot deformities The Extensor Tendon of the Big Toe (EHL) .......................8 under the supervision of Keith Baldwin, MD, MSPT, MPH. Lengthening the Tibialis Posterior Tendon .......................9 Care After Surgery .................................................10 Notes ..................................................................12 Pre-operative right foot. Post-operative -

Various Outcomes of Idiopathic Grade IV Intraventricular Haemorrhage in Term Newborns at Two Years of Age. Das S1, Bhattacharya M2, Chatterjee K3, Sarkar N4, Aich B5
Bangladesh Journal of Medical Science Vol. 17 No. 02 April’18 Case report: Various outcomes of Idiopathic Grade IV Intraventricular Haemorrhage in term newborns at two years of age. Das S1, Bhattacharya M2, Chatterjee K3, Sarkar N4, Aich B5 Bangladesh Journal of Medical Science Vol. 17 No. 02 April’18. Page : 316-318 DOI: http://dx.doi.org/10.3329/bjms.v17i2.35893 Introduction: duration of NICU stay was 15 days. Intraventricular Haemorrhage (IVH) generally Case1 (corresponds to Figure1) - At 2 years of age, occurs in infants <32 weeks and/or <1500 grams. he was developing right sided spastic hemiparetic Incidence of IVH in term neonates is 3.5-5% 1,2 . cerebral palsy. Right sided limbs exhibited 50% of IVH in term neonates is primarily caused by hypertonia, brisk deep tendon reflexes, ankle clonus trauma and asphyxia; a minority of haemorrhages and persistence of cortical thumb. The child showed is caused by extension of bleed from Subdural, early hand preference, dwarfing and dyspraxia of Subarachnoid and Intraparenchymal haemorrhage affected limbs. Electroencephalogram (EEG), Visual or caused by vascular lesions, coagulopathies or Evoked Potential (VEP), Brainstem Auditory Evoked tumours. 25% of cases have no significant risk Response (BAER) and Fundoscopy were normal factors. Most of the germinal matrix has regressed at 2 years of age. Developmental assesment was by term, so most haemorrhages (35%) arise from the done by Developmental Assesment Scale For Indian posterior tufts at the glomus in choroid plexus, 24% Infants (DASII) which is based on Bayley Scale of from Thalamus, 17% from residual Germinal Matrix Infant Development (BSID) II norms. -

Mid-Trimester Preterm Premature Rupture of Membranes (PPROM): Etiology, Diagnosis, Classification, International Recommendations of Treatment Options and Outcome
J. Perinat. Med. 2018; 46(5): 465–488 Review article Open Access Michael Tchirikov*, Natalia Schlabritz-Loutsevitch, James Maher, Jörg Buchmann, Yuri Naberezhnev, Andreas S. Winarno and Gregor Seliger Mid-trimester preterm premature rupture of membranes (PPROM): etiology, diagnosis, classification, international recommendations of treatment options and outcome DOI 10.1515/jpm-2017-0027 neonates delivered without antecedent PPROM. The “high Received January 23, 2017. Accepted May 19, 2017. Previously pub- PPROM” syndrome is defined as a defect of the chorio- lished online July 15, 2017. amniotic membranes, which is not located over the inter- nal cervical os. It may be associated with either a normal Abstract: Mid-trimester preterm premature rupture of mem- or reduced amount of amniotic fluid. It may explain why branes (PPROM), defined as rupture of fetal membranes sensitive biochemical tests such as the Amniosure (PAMG-1) prior to 28 weeks of gestation, complicates approximately or IGFBP-1/alpha fetoprotein test can have a positive result 0.4%–0.7% of all pregnancies. This condition is associ- without other signs of overt ROM such as fluid leakage with ated with a very high neonatal mortality rate as well as an Valsalva. The membrane defect following fetoscopy also increased risk of long- and short-term severe neonatal mor- fulfils the criteria for “high PPROM” syndrome. In some bidity. The causes of the mid-trimester PPROM are multi- cases, the rupture of only one membrane – either the cho- factorial. Altered membrane morphology including marked rionic or amniotic membrane, resulting in “pre-PPROM” swelling and disruption of the collagen network which is could precede “classic PPROM” or “high PPROM”. -

Hereditary Spastic Paraplegias
Hereditary Spastic Paraplegias Authors: Doctors Enza Maria Valente1 and Marco Seri2 Creation date: January 2003 Update: April 2004 Scientific Editor: Doctor Franco Taroni 1Neurogenetics Istituto CSS Mendel, Viale Regina Margherita 261, 00198 Roma, Italy. e.valente@css- mendel.it 2Dipartimento di Medicina Interna, Cardioangiologia ed Epatologia, Università degli studi di Bologna, Laboratorio di Genetica Medica, Policlinico S.Orsola-Malpighi, Via Massarenti 9, 40138 Bologna, Italy.mailto:[email protected] Abstract Keywords Disease name and synonyms Definition Classification Differential diagnosis Prevalence Clinical description Management including treatment Diagnostic methods Etiology Genetic counseling Antenatal diagnosis References Abstract Hereditary spastic paraplegias (HSP) comprise a genetically and clinically heterogeneous group of neurodegenerative disorders characterized by progressive spasticity and hyperreflexia of the lower limbs. Clinically, HSPs can be divided into two main groups: pure and complex forms. Pure HSPs are characterized by slowly progressive lower extremity spasticity and weakness, often associated with hypertonic urinary disturbances, mild reduction of lower extremity vibration sense, and, occasionally, of joint position sensation. Complex HSP forms are characterized by the presence of additional neurological or non-neurological features. Pure HSP is estimated to affect 9.6 individuals in 100.000. HSP may be inherited as an autosomal dominant, autosomal recessive or X-linked recessive trait, and multiple recessive and dominant forms exist. The majority of reported families (70-80%) displays autosomal dominant inheritance, while the remaining cases follow a recessive mode of transmission. To date, 24 different loci responsible for pure and complex HSP have been mapped. Despite the large and increasing number of HSP loci mapped, only 9 autosomal and 2 X-linked genes have been so far identified, and a clear genetic basis for most forms of HSP remains to be elucidated. -
IUGR: Intrauterine Growth Restriction
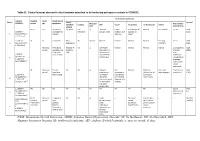
Table S1. Clinical features observed in the 6 patients described so far harboring pathogenic variants in FOXRED1. Evolutionary symptoms Variants Prenatal Onset Onset clinical Patient Lactic/ Survival FOXRED1 period age symptoms Muscular Psychomotor Metabolic Epilepsy MRI Visual Respiratory Cardiovascular Others tone development acidosis IUGR 2m Hypotonia Yes (+++) Yes ↓ Normal Latent Bronchospasm Normal AEP normal IQ: 48 Alive c.920G>A Development refractary (2m,4y,7y3m) strabismus of episodes in (15y) 1 (p.Gly307Glu) / al delay right eye infant c.733+1G>A c.920G>A NI 4y Clumsiness With No Normal Normal Normal Normal Normal Learning IQ: 99 Alive 2 (p.Gly307Glu) / exercise (+) disorders (19y) c.733+1G>A - Neonatal Premature; No (only ↑ Yes ↓ Decreased Normal Normal Normal Normal Gradually loss Alive period Hypoglycemia lactate in attenuation in of motor (22y)) Congenital LCR) the putamen skills; c.694C>T lactic acidosis and cerebellar wheelchair; (p.Gln232X) / 3 atrophy (6y) no expressive c.1289A>G language; 18 (p.Asn430Ser) understands simple commands NI Neonatal Truncal Yes Yes ↓ Delayed Eye Normal Mild non- Persistent Psychomotor Alive period hypotonia myelination movements obstructive left hepatomegaly retardation (10y) c.1054 C>T Poor feeding ventricular have always ventricular (p.Arg352Trp) / dilatation; been roving hypertrophy 4 c.1054 C>T abnormal signal bilateral optic (p.Arg352Trp) 19 in the thalami atrophy and basal ganglia (8m) c.1308G>A ND ND ND ND Yes NA NA NA NA NA NA Severe Alive (p.Val421Met) / psychomotor (¿) 5 c.1308G>A retardation (p.Val421Met)20 IUGR; Neonatal Congenital Yes Yes ↓ Large -- Persistent Dilated right - - Death (3 c.612_615dupA Cerebral period lactic periventricular severe ventricle and months) CTG intraventric acidosis. -

Cerebral Hypotonia by Mihee Bay MD (Dr
Cerebral hypotonia By Mihee Bay MD (Dr. Bay of Kennedy Krieger Institute and Johns Hopkins School of Medicine has no relevant financial relationships to disclose.) Originally released July 12, 2006; last updated February 1, 2016; expires February 1, 2019 Introduction This article includes discussion of cerebral hypotonia, central hypotonia, essential hypotonia, benign congenital hypotonia, and floppy infant. The foregoing terms may include synonyms, similar disorders, variations in usage, and abbreviations. Overview Hypotonia is a clinical manifestation of numerous diseases affecting the central and/or peripheral motor nervous system. The key to accurate diagnosis involves integral steps of evaluation that include a detailed history, examination, and diagnostic tests. “Cerebral” (or central) hypotonia implies pathogenesis from abnormalities from the central nervous system, and related causal disorders include cerebral dysgenesis and genetic or metabolic disorders. Patients with central hypotonia generally have hypotonia without associated weakness, in contrast to the peripheral (lower motor neuron) causes, which typically produce both hypotonia and muscle weakness. Hypotonia is a clinical manifestation of over 500 genetic disorders; thus, a logical, stepwise approach to diagnosis is essential. With recent advances in the field of genetic testing, diagnostic yield will undoubtedly improve. There is no cure, but treatment includes supportive therapies, such as physical and occupational therapy, and diagnosis-specific management. Key points • Hypotonia is reduced tension or resistance of passive range of motion. • The first step in the evaluation of a child with hypotonia is localization to the central (“cerebral”) or peripheral nervous system, or both. • Central hypotonia is more likely to be noted axially with normal strength and hyperactive to normal deep tendon reflexes.