Flora Aparecida Milton
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alpha Actinin 4: an Intergral Component of Transcriptional
ALPHA ACTININ 4: AN INTERGRAL COMPONENT OF TRANSCRIPTIONAL PROGRAM REGULATED BY NUCLEAR HORMONE RECEPTORS By SIMRAN KHURANA Submitted in partial fulfillment of the requirements for the degree of doctor of philosophy Thesis Advisor: Dr. Hung-Ying Kao Department of Biochemistry CASE WESTERN RESERVE UNIVERSITY August, 2011 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of SIMRAN KHURANA ______________________________________________________ PhD candidate for the ________________________________degree *. Dr. David Samols (signed)_______________________________________________ (chair of the committee) Dr. Hung-Ying Kao ________________________________________________ Dr. Edward Stavnezer ________________________________________________ Dr. Leslie Bruggeman ________________________________________________ Dr. Colleen Croniger ________________________________________________ ________________________________________________ May 2011 (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. TABLE OF CONTENTS LIST OF TABLES vii LIST OF FIGURES viii ACKNOWLEDEMENTS xii LIST OF ABBREVIATIONS xiii ABSTRACT 1 CHAPTER 1: INTRODUCTION Family of Nuclear Receptors 3 Mechanism of transcriptional regulation by co-repressors and co-activators 8 Importance of LXXLL motif of co-activators in NR mediated transcription 12 Cyclic recruitment of co-regulators on the target promoters 15 Actin and actin related proteins (ABPs) in transcription -

Role of Nuclear Receptors in Central Nervous System Development and Associated Diseases
Role of Nuclear Receptors in Central Nervous System Development and Associated Diseases The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Olivares, Ana Maria, Oscar Andrés Moreno-Ramos, and Neena B. Haider. 2015. “Role of Nuclear Receptors in Central Nervous System Development and Associated Diseases.” Journal of Experimental Neuroscience 9 (Suppl 2): 93-121. doi:10.4137/JEN.S25480. http:// dx.doi.org/10.4137/JEN.S25480. Published Version doi:10.4137/JEN.S25480 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:27320246 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Journal name: Journal of Experimental Neuroscience Journal type: Review Year: 2015 Volume: 9(S2) Role of Nuclear Receptors in Central Nervous System Running head verso: Olivares et al Development and Associated Diseases Running head recto: Nuclear receptors development and associated diseases Supplementary Issue: Molecular and Cellular Mechanisms of Neurodegeneration Ana Maria Olivares1, Oscar Andrés Moreno-Ramos2 and Neena B. Haider1 1Department of Ophthalmology, Schepens Eye Research Institute, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, USA. 2Departamento de Ciencias Biológicas, Facultad de Ciencias, Universidad de los Andes, Bogotá, Colombia. ABSTRACT: The nuclear hormone receptor (NHR) superfamily is composed of a wide range of receptors involved in a myriad of important biological processes, including development, growth, metabolism, and maintenance. -

Full-Text PDF (Final Published Version)
Alexander, S. P. H., Cidlowski, J. A., Kelly, E., Marrion, N. V., Peters, J. A., Faccenda, E., Harding, S. D., Pawson, A. J., Sharman, J. L., Southan, C., Davies, J. A., & CGTP Collaborators (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174, S208-S224. https://doi.org/10.1111/bph.13880 Publisher's PDF, also known as Version of record License (if available): CC BY Link to published version (if available): 10.1111/bph.13880 Link to publication record in Explore Bristol Research PDF-document This is the final published version of the article (version of record). It first appeared online via Wiley at https://doi.org/10.1111/bph.13880 . Please refer to any applicable terms of use of the publisher. University of Bristol - Explore Bristol Research General rights This document is made available in accordance with publisher policies. Please cite only the published version using the reference above. Full terms of use are available: http://www.bristol.ac.uk/red/research-policy/pure/user-guides/ebr-terms/ S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology (2017) 174, S208–S224 THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Nuclear hormone receptors Stephen PH Alexander1, John A Cidlowski2, Eamonn Kelly3, Neil V Marrion3, John A Peters4, Elena Faccenda5, Simon D Harding5,AdamJPawson5, Joanna L Sharman5, Christopher Southan5, Jamie A Davies5 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical -

Alternative Splicing in the Nuclear Receptor Superfamily Expands Gene Function to Refine Endo-Xenobiotic Metabolism S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2020/01/24/dmd.119.089102.DC1 1521-009X/48/4/272–287$35.00 https://doi.org/10.1124/dmd.119.089102 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 48:272–287, April 2020 Copyright ª 2020 by The American Society for Pharmacology and Experimental Therapeutics Minireview Alternative Splicing in the Nuclear Receptor Superfamily Expands Gene Function to Refine Endo-Xenobiotic Metabolism s Andrew J. Annalora, Craig B. Marcus, and Patrick L. Iversen Department of Environmental and Molecular Toxicology, Oregon State University, Corvallis, Oregon (A.J.A., C.B.M., P.L.I.) and United States Army Research Institute for Infectious Disease, Frederick, Maryland (P.L.I.) Received August 16, 2019; accepted December 31, 2019 ABSTRACT Downloaded from The human genome encodes 48 nuclear receptor (NR) genes, whose Exon inclusion options are differentially distributed across NR translated products transform chemical signals from endo- subfamilies, suggesting group-specific conservation of resilient func- xenobiotics into pleotropic RNA transcriptional profiles that refine tionalities. A deeper understanding of this transcriptional plasticity drug metabolism. This review describes the remarkable diversifica- expands our understanding of how chemical signals are refined and tion of the 48 human NR genes, which are potentially processed into mediated by NR genes. This expanded view of the NR transcriptome over 1000 distinct mRNA transcripts by alternative splicing (AS). The informs new models of chemical toxicity, disease diagnostics, and dmd.aspetjournals.org average human NR expresses ∼21 transcripts per gene and is precision-based approaches to personalized medicine. -

WO 2019/089982 Al 09 May 2019 (09.05.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2019/089982 Al 09 May 2019 (09.05.2019) W 1P O PCT (51) International Patent Classification: (71) Applicant: JUNO THERAPEUTICS, INC. [US/US]; 400 C12Q 1/6897 (2018.01) CI2N 15/65 (2006.01) Dexter Avenue N, Suite 1200, Seattle, Washington 98109 C12Q 1/02 (2006.01) G01N 33/50 (2006.01) (US). C12N 5/0783 (2010.01) (72) Inventors: AMIN, Rupesh; 400 Dexter Avenue N., Suite (21) International Application Number: 1200, Seattle, Washington 98109 (US). CHEN, Aye; 400 PCT/US20 18/058781 Dexter Avenue N., Suite 1200, Seattle, Washington 98109 (US). (22) International Filing Date: 0 1 November 2018 (01. 11.2018) (74) Agent: AHN, Sejin et al.; Morrison & Foerster LLP, 1253 1 High Bluff Drive, Suite 100, San Diego, California (25) Filing Language: English 92130-2040 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/580,405 0 1 November 2017 (01. 11.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/596,758 08 December 2017 (08. 12.2017) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, 62/599,672 15 December 2017 (15. 12.2017) US DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, (54) Title: METHOD OF ASSESSING ACTIVITY OF RECOMBINANT ANTIGEN RECEPTORS FIG. -

Effects of Maternal Undernutrition During Lactation on Aromatase, Estrogen, and Androgen Receptors Expression in Rat Testis at Weaning
301 Effects of maternal undernutrition during lactation on aromatase, estrogen, and androgen receptors expression in rat testis at weaning Cı´ntia Vilanova Teixeira, Dorothe´e Silandre1, Alba Marcelly de Souza Santos, Christelle Delalande1, Francisco J B Sampaio, Serge Carreau1 and Cristiane da Fonte Ramos Urogenital Research Unit, State University of Rio de Janeiro, Rio de Janeiro, RJ, Brazil 1Biochemistry, INRA2006-EA2608, University, Esplanade de la Paix, 14032, CAEN-Cedex, France (Requests for offprints should be addressed to S Carreau; Email: [email protected]) (C da Fonte Ramos is in US postdoctoral position at Stanford University, Department of Urology, California, USA). Abstract The goal of this study was to evaluate the effects of maternal concentration (ng/ml) was significantly higher in the PER malnutrition during lactation on serum levels of testosterone group compared with ER and C groups (CZ0.09G0.012; and estradiol, testicular testosterone concentration, aromatase, PERZ0.45G0.04; ERZ0.15G0.03, P!0.01). Testicular testicular androgen (AR) and estrogen a (ERa) receptors testosterone concentration (CZ2.1G0.43; PERZ6.5G0.7; expression in the pups at weaning. From parturition until ERZ13G2.3, P!0.01) was increased in treated groups weaning, Wistar rats were separated into three groups: (C) when compared with controls. The estradiol serum concen- control group, with free access to a standard laboratory diet tration (pg/ml) was lower in both dietary groups (CZ74G containing 23% protein; protein-energy restricted (PER) 4.6; PERZ49G3.2; ERZ60G5.5, P!0.01). The amounts group, with free access to an isoenergy and protein-restricted of aromatase mRNA and ERa transcripts were significantly diet containing 8% protein; and energy-restricted (ER) lower (P!0.05) in PER and ER groups; conversely AR (both group, receiving standard laboratory diet in restricted mRNA and protein) was significantly enhanced (P!0.05) in quantities, which were calculated according to the mean treated animals. -

Rs 000-000-260 AKT/Pkba Substrate 1.0 Mg/Ml 500 Μ
Catalog Number Display Name Concentration ValueSize Default Unit List price - Rs 000-000-260 AKT/PKBa Substrate 1.0 mg/mL 500 µg 24,434.00 000-000-264 NFKB p65 (Rel A) pS276 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-264NP NFKB p65 (Rel A) S276 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-266 p65 (Rel A) pS529 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-266NP p65 (Rel A) S529 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-383 DYKDDDDK (FLAG®) Peptide 1 mg/mL 1.0 mg 12,285.00 000-000-398 ATM S1981 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-400 ATM pS1981 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-401 AKT Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-402 Angiopoietin 2 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-403 Angiopoietin 1 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-404 Osteopontin Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-405 NOTCH 1 (intra) (Human specific) Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-407 NOTCH 1 (Human specific) Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-408 NOTCH 2 (Human specific) Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-410 ASK-1 phospho specific pS83 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-410NP ASK-1 non phospho specific S83 Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-433 Huntington pS421 Control Phospho Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-450 Huntington S421 Control Non-Phospho Peptide 1.0 mg/mL 50 µg 13,787.00 000-000-H47 Triple FLAG® Peptide 1.0 mg/ml 1.0 mg 12,285.00 000-000-K95 Beta Amyloid 40, Peptide 1.0 mg/mL 1.0 mg 1,06,334.00 000-000-K95S Beta Amyloid 40, Peptide 1.0 mg/mL 0.5 mg 27,164.00 000-000-M33 LL-37, Rhodamine -

Binding Characteristics of Estrogen Receptor (ER) in Atlantic Croaker
BIOLOGY OF REPRODUCTION 61, 51±60 (1999) Binding Characteristics of Estrogen Receptor (ER) in Atlantic Croaker (Micropogonias undulatus) Testis: Different Af®nity for Estrogens and Xenobiotics from that of Hepatic ER 1 Anna Katrina Loomis and Peter Thomas2 Department of Marine Science, Marine Science Institute, University of Texas at Austin, Port Aransas, Texas 78373 ABSTRACT spermatogenesis and fertility [10]. However, although the existence of estrogens and ERs in males is well recognized An estrogen receptor (ER) was identi®ed in cytosolic and nu- throughout the vertebrates, their precise physiological roles Downloaded from https://academic.oup.com/biolreprod/article/61/1/51/2734627 by guest on 31 December 2020 clear fractions of the testis in a marine teleost, Atlantic croaker in male reproduction remain unclear. (Micropogonias undulatus). A single class of high af®nity, low capacity, and displaceable binding sites was identi®ed by satu- The ER in the testis is also a potential target for endo- ration analysis, with a K of 0.40 nM in cytosolic extracts and a crine disruption by xenoestrogens. Evidence is accumulat- d ing which suggests that estrogenic compounds, including Kd of 0.33 nM in nuclear extracts. Competition studies demon- strated that the receptor was highly speci®c for estrogens (di- pesticides and industrial chemicals, impair male reproduc- ethylstilbestrol . estradiol k estriol 5 estrone) and also bound tive function in wildlife [11, 12]. Rainbow trout exposed to several antiestrogens. Testosterone and 5a-dihydrotestosterone estrogenic alkylphenolic chemicalsÐnonionic surfactants had much lower af®nities for the receptor, whereas no displace- that are often detected in sewage ef¯uentÐexhibit inhibi- ment of speci®c binding occurred with 11-ketotestosterone or tion of testicular growth [13]. -

Exosome-Derived Micrornas of Human Milk and Their Effects on Infant Health and Development
biomolecules Review Exosome-Derived MicroRNAs of Human Milk and Their Effects on Infant Health and Development Bodo C. Melnik 1,* , Wolfgang Stremmel 2, Ralf Weiskirchen 3 , Swen Malte John 1,4 and Gerd Schmitz 5 1 Department of Dermatology, Environmental Medicine and Health Theory, University of Osnabrück, D-49076 Osnabrück, Germany; [email protected] 2 Private Praxis for Internal Medicine, Beethovenstraße 2, D-76530 Baden-Baden, Germany; [email protected] 3 Institute of Molecular Pathobiochemistry, Experimental Gene Therapy and Clinical Chemistry (IFMPEGKC), RWTH University Hospital Aachen, D-52074 Aachen, Germany; [email protected] 4 Institute for Interdisciplinary Dermatological Prevention and Rehabilitation (iDerm), University of Osnabrück, D-49076 Osnabrück, Germany 5 Institute for Clinical Chemistry and Laboratory Medicine, University Hospital of Regensburg, University of Regensburg, D-93053 Regensburg, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-5241-988060 Abstract: Multiple biologically active components of human milk support infant growth, health and development. Milk provides a wide spectrum of mammary epithelial cell-derived extracellular vesicles (MEVs) for the infant. Although the whole spectrum of MEVs appears to be of functional importance for the growing infant, the majority of recent studies report on the MEV subfraction of milk exosomes (MEX) and their miRNA cargo, which are in the focus of this review. MEX and the dominant miRNA-148a play a key role in intestinal maturation, barrier function and suppression of nuclear factor-κB (NF-κB) signaling and may thus be helpful for the prevention and treatment of Citation: Melnik, B.C.; Stremmel, W.; necrotizing enterocolitis. MEX and their miRNAs reach the systemic circulation and may impact Weiskirchen, R.; John, S.M.; Schmitz, epigenetic programming of various organs including the liver, thymus, brain, pancreatic islets, beige, G. -
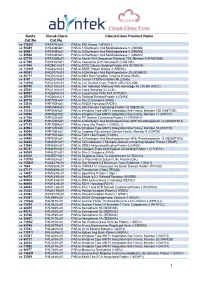
1 Santa Cat.No Cloud-Clone Cat.No. Cloud-Clone Product Name
Santa Cloud -Clone Cloud -Clone Product Name Cat.No Cat.No. sc -130253 PAS477Hu01 PAB to RIO Kinase 1 (RIOK1) sc -50485 PAS446Ra01 PAB to A Disintegrin And Metalloprotease 5 (ADAM5) sc -50487 PAS445Ra01 PAB to A Disintegrin And Metalloprotease 6 (ADAM6) sc -25588 PAS444Ra01 PAB to A Disintegrin And Metalloprotease 1 (ADAM1) sc -87719 PAR758Mu01 PAB to Family With Sequence Similarity 135, Member B (FAM135B) sc -67296 PAR493Hu01 PAB to Coenzyme Q10 Homolog B (COQ10B) sc -67048 PAQ981Hu01 PAB to S100 Calcium Binding Protein A15 (S100A15) sc -130269 PAQ342Hu01 PAB to SRSF Protein Kinase 3 (SRPK3) sc -98582 PAQ207Hu01 PAB to A Disintegrin And Metalloprotease 20 (ADAM20) sc -20711 PAQ164Hu01 PAB to BMX Non Receptor Tyrosine Kinase (BMX) sc -9147 PAQ127Hu01 PAB to Cluster Of Differentiation 8b (CD8b) sc -130184 PAQ124Hu01 PAB to Cell Division Cycle Protein 25B (CDC25B) sc -98790 PAQ118Hu01 PAB to Cell Adhesion Molecule With Homology To L1CAM (CHL1) sc -25361 PAQ116Hu01 PAB to Clock Homolog (CLOCK) sc -98937 PAQ089Hu01 PAB to Cytochrome P450 3A7 (CYP3A7) sc -25519 PAQ088Hu01 PAB to Dickkopf Related Protein 4 (DKK4) sc -28778 PAP797Hu01 PAB to Pim-2 Oncogene (PIM2) sc -33626 PAP750Hu01 PAB to RAD51 Homolog (RAD51) sc -8333 PAP694Mu01 PAB to SH2 Domain Containing Protein 1A (SH2D1A) sc -25524 PAP553Hu01 PAB to Wingless Type MMTV Integration Site Family, Member 10B (WNT10B) sc -50360 PAP552Hu01 PAB to Wingless Type MMTV Integration Site Family, Member 11 (WNT11) sc -87368 PAP332Hu01 PAB to PR Domain Containing Protein 14 (PRDM14) sc -25583 PAP226Hu01 -

A SILAC-Based DNA Protein Interaction Screen That Identifies Candidate Binding Proteins to Functional
Downloaded from genome.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements Gerhard Mittler1,2,4, Falk Butter3 and Matthias Mann3 1 Center for Experimental Bioinformatics (CEBI), University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark 2 Current address: Dept. Cellular and Molecular Immunology, Proteomics, Max-Planck Institute of Immunobiology, Stübeweg 51, D-79108 Freiburg, Germany 3 Dept. Proteomics and Signal Transduction, Max-Planck-Institute for Biochemistry, Am Klopferspitz 18, D-82152 Martinsried, Germany 4 BIOSS – Center of Biological Signalling Studies, Albert-Ludwigs-University Freiburg, Albertstrassse 19, D-79104 Freiburg, Germany Running title: SILAC for DNA-binding proteins Downloaded from genome.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Determining the underlying logic that governs the networks of gene expression in higher eukaryotes is an important task in the post-genome era. Sequence-specific transcription factors (TFs) that can read the genetic regulatory information and proteins that interpret the information provided by CpG methylation are crucial components of the system that controls the transcription of protein-coding genes by RNA polymerase II. We have previously described Stable Isotope Labeling by Amino acids in Cell culture (SILAC) for the quantitative comparison of proteomes and the determination of protein-protein interactions. Here, we report a generic and scalable strategy to uncover such DNA protein interactions by SILAC that uses a fast and simple one-step affinity capture of transcription factors from crude nuclear extracts. -

Gastroenterologie, Hepatologie, Infektionskrankheiten
Aus der Medizinischen Universitätsklinik und Poliklinik Tübingen Abteilung Innere Medizin I Schwerpunkt: Gastroenterologie, Hepatologie, Infektionskrankheiten Überprüfung von Merbromin und Eosin Y als potenzielle Zellregeneration fördernde Substanzen Inaugural-Dissertation zur Erlangung des Doktorgrades der Medizin der Medizinischen Fakultät der Eberhard Karls Universität zu Tübingen vorgelegt von Bajwa, geb. Sindhu, Bariya Ahmed 2019 Dekan: Professor Dr. I. B. Autenrieth 1. Berichterstatter: Professor Dr. N.P. Malek 2. Berichterstatter: Professor Dr. A. Hartkopf Tag der Disputation: 10.07.2019 2 Danksagung An dieser Stelle möchte ich meinen besonderen Dank nachstehenden Personen entgegen bringen, ohne deren Mithilfe die Anfertigung dieser Promotionsschrift niemals zustande gekommen wäre: Mein Dank gilt zunächst Prof. Malek, meinem Doktorvater, für die Betreuung dieser Arbeit, für die vielen Denkanstöße und Ideen, die mir einen kritischen Zugang zu dieser Thematik eröffneten und für die sowohl wissenschaftliche als auch klinische Unterstützung meiner Laufbahn. Ferner möchte ich Dr. Przemyslaw Bozko für die Mitbetreuung an meiner Dissertation und für die freundliche Hilfe danken. Ich danke außerdem Labormitglied Tim Scholta für die stetige Hilfsbereitschaft während meiner wissenschaftlichen Tätigkeit. Tief verbunden und dankbar bin ich meiner Familie und insbesondere meinem Ehemann, Asad Hameed Bajwa, für seine unglaublich hilfreiche Unterstützung in Form von Motivation, als mir diese gefehlt hat, für seine Geduld, und sein Verständnis