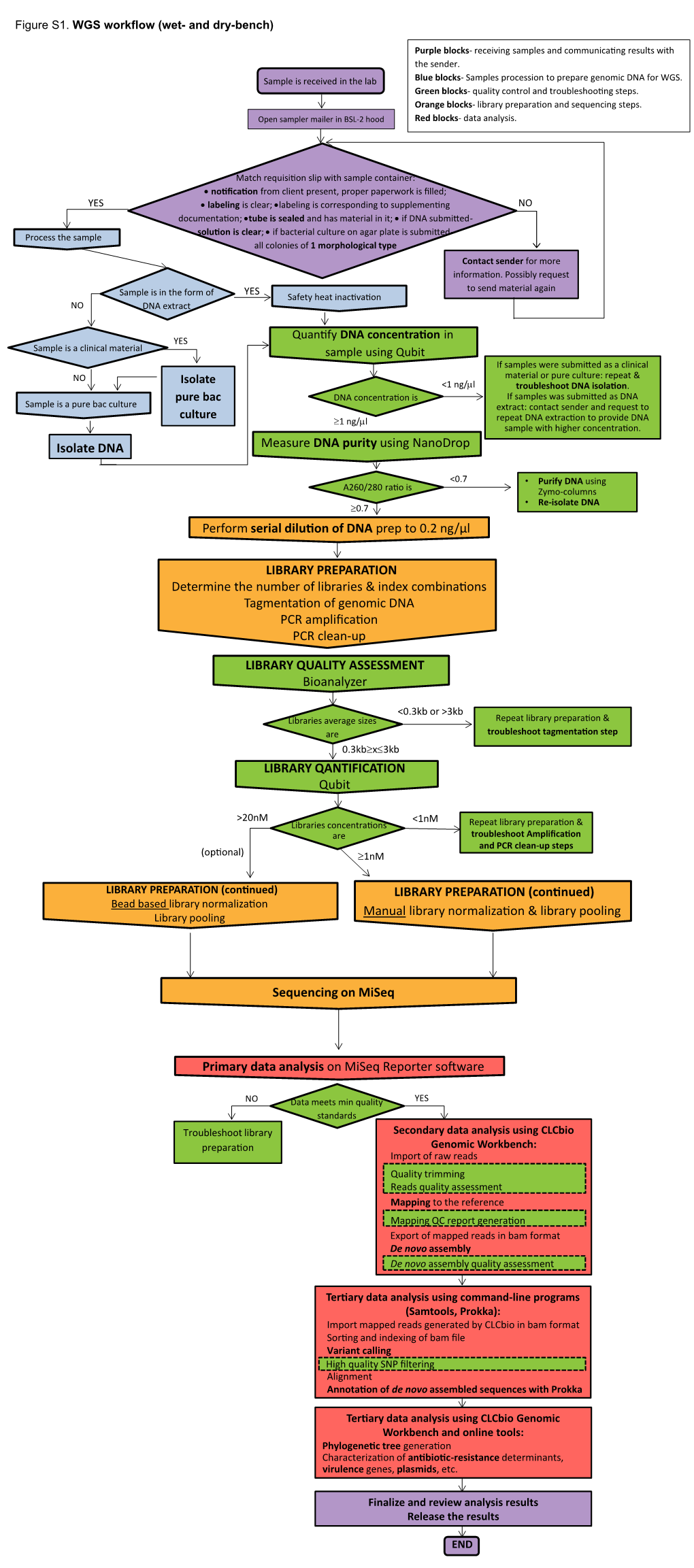
Perform Serial Dilution of DNA Prep to 0.2 Ng/Μl Isolate DNA
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Using Serial Dilution to Understand Ppm/Ppb
Using Serial Dilution to Understand ppm/ppb Adapted from Investigating Groundwater: The Fruitvale Story, Science Education for Public Understanding Program (SEPUP), Lawrence Hall of Science (1996) by Dana Haine, UNC Superfund Research Program. Overview: Students will perform a serial dilution of food coloring to create highly diluted solutions to learn about the units, parts per million (ppm) and parts per billion (ppb), that scientists often use to describe chemical contamination of water or soil. Objectives: At the end of this activity, students will be able to: • Define serial dilution; • Define ppm and ppb; • Observe that a contaminant can be present in water even if it isn’t visible. Alignment to North Carolina Essential Standards for Science This lesson addresses components of the specific learning objectives: 8th Grade Science 8.E.1.3 Predict the safety and potability of water supplies in North Carolina based on physical and biological factors, including temperature, dissolved oxygen, pH, nitrates and phosphates, turbidity, bio-indicators 8.E.1.4 Conclude that the good health of humans requires: monitoring of the hydrosphere, water quality standards, methods of water treatment, maintaining safe water quality, stewardship Earth and Environmental Science EEn.2.4.1 Evaluate human influences on freshwater availability. EEn.2.4.2 Evaluate human influences on water quality in North Carolina’s river basins, wetlands and tidal environments. Physical Science PSc.2.1.2 Explain the phases of matter and the physical changes that matter undergoes. Materials: Student Data Collection Sheet, one per student Plastic tray with at least 9 wells or 9 test tubes, one tray/set of tubes per group White scrap paper to place under trays/tubes to observe results, one per group Red Food Coloring Two small cups of water, one labeled “rinse” Medicine dropper, one per group Paper towels Red colored pencils (optional) Duration 20-30 minutes Procedure: 1. -

Laboratory Exercises in Microbiology: Discovering the Unseen World Through Hands-On Investigation
City University of New York (CUNY) CUNY Academic Works Open Educational Resources Queensborough Community College 2016 Laboratory Exercises in Microbiology: Discovering the Unseen World Through Hands-On Investigation Joan Petersen CUNY Queensborough Community College Susan McLaughlin CUNY Queensborough Community College How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/qb_oers/16 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] Laboratory Exercises in Microbiology: Discovering the Unseen World through Hands-On Investigation By Dr. Susan McLaughlin & Dr. Joan Petersen Queensborough Community College Laboratory Exercises in Microbiology: Discovering the Unseen World through Hands-On Investigation Table of Contents Preface………………………………………………………………………………………i Acknowledgments…………………………………………………………………………..ii Microbiology Lab Safety Instructions…………………………………………………...... iii Lab 1. Introduction to Microscopy and Diversity of Cell Types……………………......... 1 Lab 2. Introduction to Aseptic Techniques and Growth Media………………………...... 19 Lab 3. Preparation of Bacterial Smears and Introduction to Staining…………………...... 37 Lab 4. Acid fast and Endospore Staining……………………………………………......... 49 Lab 5. Metabolic Activities of Bacteria…………………………………………….…....... 59 Lab 6. Dichotomous Keys……………………………………………………………......... 77 Lab 7. The Effect of Physical Factors on Microbial Growth……………………………... 85 Lab 8. Chemical Control of Microbial Growth—Disinfectants and Antibiotics…………. 99 Lab 9. The Microbiology of Milk and Food………………………………………………. 111 Lab 10. The Eukaryotes………………………………………………………………........ 123 Lab 11. Clinical Microbiology I; Anaerobic pathogens; Vectors of Infectious Disease….. 141 Lab 12. Clinical Microbiology II—Immunology and the Biolog System………………… 153 Lab 13. Putting it all Together: Case Studies in Microbiology…………………………… 163 Appendix I. -

Determination of the Solubility Product of an Ionic Compound
Determination of the Solubility Product of an Ionic Compound Kyle Miller January 11, 2007 1 Data The following data were collected. Dilution Precipitation well Ca2+ #6 OH− #4 2 Calculations 2.1 Calcium Ion Serial Dilution The first well without a precipitate is well number 6. The concentration of calcium ions per well is 0.10 [Ca2+] = M (1) 2n where n is the well number. So, the concentration of calcium ions in well number 6 is 0.10 −3 26 M = 1.6 × 10 M For each of the wells in this series, the concentration of added NaOH was a constant 0.1 M 2 = 0.05 M after being added to each well. 2+ − The Ksp for this ion equilibrium, Ca(OH)2 Ca + 2OH , is 2+ −2 Ksp = Ca OH (2) So, putting in the concentrations for well number 6, which is assumed to have complete −3 2 −6 dissociation of the calcium hydroxide, Ksp = 1.6 × 10 [0.05] = 3.9 × 10 1 2.2 Hydroxide Ion Serial Dilution The first well without precipitate for this series is well number 4. The concentration of hydroxide ions per well is 0.10 [OH−] = M (3) 2n again, with n being the well number. So, the concentration of hydroxide ions in well 0.10 −3 number 4 is 24 M = 6.3 × 10 M For each of the wells in this series, the concentration of added calcium nitrate was a constant 0.1 M 2 = 0.05 M after being added to each well. Using the ion equilibrium equation and these concentrations, we can find that Ksp = [0.05] 6.3 × 10−32 = 2.0 × 10−6 ¯ 3.9×10−6+2.0×10−6 −6 Then, Ksp = 2 = 3.0 × 10 −6 The actual Ksp is 5.02×10 and the percent error from this value to the calculated average |3.0×10−6−5.02×10−6| is 5.02×10−6 = 40.% 3 Discussion 1. -

Experiment 16
FV 2/10/2017 Experiment 16 HYDRONIUM ION CONCENTRATION MATERIALS: 7 centrifuge tubes with 10 mL mark, six 25 x 150 mm large test tubes, 5 mL pipet, 50 mL beaker, 50 mL graduated cylinder, 1.0 M HCl, 1.0 M CH3COOH, CH3COONa, methyl violet and methyl orange indicator solutions, test tube rack, calibrated pH meter (instructor use only). PURPOSE: The purpose of this experiment is to perform serial dilutions and use indicators to estimate the pH of various solutions. LEARNING OBJECTIVES: By the end of this experiment, the students should be able to demonstrate the following proficiencies: 1. Prepare solutions by serial dilution. + 2. Correlate the H3O ion concentration of a solution with its pH value. 3. Use indicators to estimate the pH of solutions of various acid concentrations. 4. Explain the common ion effect. 5. Calculate percent dissociation of a weak acid. PRE-LAB: Read over the experiment and complete the pre-lab questions on OWL before the lab. DISCUSSION: In any aqueous solution, the product of the hydronium ion concentration and the hydroxide ion concentration is equal to a constant, known as Kw. At a temperature of 25°C, this product is: + - -14 Kw = [H3O ]⋅ [OH ] = 1.0 x 10 (25° C) + - If the [H3O ] concentration is altered, the [OH ] concentration changes so that the product of the two terms remains 1.0 x 10-14. In pure, distilled water at 25°C: + - -7 [H3O ] = [OH ] = 1.0 x 10 M (25° C) To describe acidity in a simple way, the pH scale was adopted: + pH = -log[H3O ] Thus, for pure, distilled water at 25°C: -7 = ° pH = -log(1.0 x 10 ) 7.00 (25 C) + The practical range of pH in aqueous solutions at 25°C is from 0 to 14. -

Robust Estimation of Bacterial Cell Count from Optical Density
bioRxiv preprint first posted online Oct. 13, 2019; doi: http://dx.doi.org/10.1101/803239. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Robust Estimation of Bacterial Cell Count from Optical Density Jacob Beal1*, Natalie G. Farny2*, Traci Haddock-Angelli3*, Vinoo Selvarajah3, Geoff S. Baldwin4*, Russell Buckley-Taylor4, Markus Gershater5*, Daisuke Kiga6, John Marken7, Vishal Sanchania5, Abigail Sison3, Christopher T. Workman8*, and the iGEM Interlab Study Contributors 1 Raytheon BBN Technologies, Cambridge, MA, USA 2 Department of Biology and Biotechnology, Worcester Polytechnic Institute, Worcester, MA, USA 3 iGEM Foundation, Cambridge, MA, USA 4 Department of Life Sciences and IC-Centre for Synthetic Biology, Imperial College London, London, UK 5 Synthace, London, UK 6 Faculty of Science and Engineering, School of Advanced Science and Engineering, Waseda University, Tokyo, Japan 7 Department of Bioengineering, California Institute of Technology, Pasadena, CA, USA 8 DTU-Bioengineering, Technical University of Denmark, Kongens Lyngby, Denmark Membership list is provided in Supplementary Note: iGEM Interlab Study Contributors. * [email protected] (J.B.), [email protected] (N.G.F.), [email protected] (T.H-A.), [email protected] (G.S.B.), [email protected] (M.G.), [email protected] (C.T.W.) Abstract Optical density (OD) is a fast, cheap, and high-throughput measurement widely used to estimate the density of cells in liquid culture. These measurements, however, cannot be compared between instruments without a standardized calibration protocol and are challenging to relate to actual cell count. -

Standard Methods for the Examination of Water and Wastewater
Standard Methods for the Examination of Water and Wastewater Part 1000 INTRODUCTION 1010 INTRODUCTION 1010 A. Scope and Application of Methods The procedures described in these standards are intended for the examination of waters of a wide range of quality, including water suitable for domestic or industrial supplies, surface water, ground water, cooling or circulating water, boiler water, boiler feed water, treated and untreated municipal or industrial wastewater, and saline water. The unity of the fields of water supply, receiving water quality, and wastewater treatment and disposal is recognized by presenting methods of analysis for each constituent in a single section for all types of waters. An effort has been made to present methods that apply generally. Where alternative methods are necessary for samples of different composition, the basis for selecting the most appropriate method is presented as clearly as possible. However, samples with extreme concentrations or otherwise unusual compositions or characteristics may present difficulties that preclude the direct use of these methods. Hence, some modification of a procedure may be necessary in specific instances. Whenever a procedure is modified, the analyst should state plainly the nature of modification in the report of results. Certain procedures are intended for use with sludges and sediments. Here again, the effort has been to present methods of the widest possible application, but when chemical sludges or slurries or other samples of highly unusual composition are encountered, the methods of this manual may require modification or may be inappropriate. Most of the methods included here have been endorsed by regulatory agencies. Procedural modification without formal approval may be unacceptable to a regulatory body. -

Why Are DLS Measurements in High Concentration Solutions Difficult?
Why are DLS measurements in high concentration solutions difficult? Brookhaven Instruments • Holtsville, New York Dynamic Light Scattering (DLS) is an effective measurement technique used for measuring the hydrodynamic size of common nanomaterials including colloids, nanoparticles, proteins, and polymers. Despite the versatility of this technique, there are several important considerations that cannot be ignored when using light scattering to characterize high-concentration solutions. While it is possible to make measurements on high volume-fraction samples without dilution, it raises additional questions about the meaning of the hydrodynamic size. To understand why this is the case we need to discuss two effects encountered in concentrated solutions: multiple scattering and mutual diffusion. Equivalent Hydrodynamic Size The size obtained from DLS is a hydrodynamic size, which is derived from measurements of particle diffusion. All freely diffusing particles are constantly undergoing Brownian Motion in proportion to thermal energy expressed as the product of the Boltzmann constant by the temperature (kbT). A single particle would experience Brownian Diffusion in inverse proportion to its size, thus the diffusion coefficient, DT, of this particle can be used to calculate its hydrodynamic diameter, dH, as shown in the Stokes-Einstein equation: DT = kb T / 3πηdH dH can be obtained if the temperature, T, and viscosity, η, are also known. This single particle diffusion coefficient is the result of self-diffusion; therefore, in the limit of infinite dilution, this calculated size is fully equivalent to its hydrodynamic size. In the simplest case a single decay rate, Г, is extracted from the DLS autocorrelation function (ACF). This Г is the reciprocal of the characteristic relaxation time, τt, such that: g(1)( τ) = exp(-Г τ) Figure 1.0 Simulated ACF’s for decay rates of Г = 7000, 4000, and 1000 s-1 (corresponding to 24, 41, and 165 nm diameter spherical particles). -

UC Berkeley UC Berkeley Electronic Theses and Dissertations
UC Berkeley UC Berkeley Electronic Theses and Dissertations Title Fluidic Microvalve Digital Processors for Automated Biochemical Analysis Permalink https://escholarship.org/uc/item/3mg2x7gh Author Jensen, Erik Christian Publication Date 2011 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California Fluidic Microvalve Digital Processors for Automated Biochemical Analysis by Erik C. Jensen A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Biophysics in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, BERKELEY Committee in charge: Professor Richard A. Mathies, Chair Professor Lydia Sohn Professor Jay Groves Professor Bernhard Boser Spring 2011 Fluidic Microvalve Digital Processors for Automated Biochemical Analysis Copyright 2011 by Erik C. Jensen Abstract Fluidic Microvalve Digital Processors for Automated Biochemical Analysis by Erik C. Jensen Doctor of Philosophy in Biophysics University of California, Berkeley Professor Richard A. Mathies, Chair The development of microfluidic sample processing and microvalve technology offers significant, thus far unmet opportunities for the miniaturization and large scale integration of automated laboratory systems. In this dissertation, the transistor-like nature of monolithic membrane valves for control of airflow and fluid flow is exploited to develop microfluidic processors for performing diverse bioassay procedures on a common programmable microchip format. The transistor-nature of pneumatic microvalves is first exploited to fabricate devices that perform AND, OR, and NOT transistor-to-transistor logic operations. With this system, microvalves are used to control the actuation of other microvalves by regulating airflow. As a demonstration of computational universality, these operators are combined to perform more complex digital logic operations including binary addition. -

Interlab Study Introduction Reliable and Repeatable Measurement Is a Key Component to All Engineering Disciplines
Interlab study Introduction Reliable and repeatable measurement is a key component to all engineering disciplines. The same holds true for synthetic biology, which has also been called engineering biology. However, the ability to repeat measurements in different labs has been difficult. The Measurement Committee, through the InterLab study, has been developing a robust measurement procedure for green fluorescent protein (GFP) over the last several years. We chose GFP as the measurement marker for this study since it's one of the most used markers in synthetic biology and, as a result, most laboratories are equipped to measure this protein. The aim to improve the measurement tools available to both the iGEM community and the synthetic biology community as a whole. One of the big challenges in synthetic biology measurement has been that fluorescence data usually cannot be compared because it has been reported in different units or because different groups process data in different ways. Many have tried to work around this using “relative expression” comparisons; however, being unable to directly compare measurements makes it harder to debug engineered biological constructs, harder to effectively share constructs between labs, and harder even to just interpret your experimental controls. The InterLab protocol aims to address these issues by providing researchers with a detailed protocol and data analysis form that yields absolute units for measurements of GFP in a plate reader. Goal for the Fifth InterLab The goal of the iGEM InterLab Study is to identify and correct the sources of systematic variability in synthetic biology measurements, so that eventually, measurements that are taken in different labs will be no more variable than measurements taken within the same lab. -

I. Organics (Bod, Cod, Toc, O&G)
Understanding Laboratory Wastewater Tests: I. ORGANICS (BOD, COD, TOC, O&G) Since the implementation of the Clean Water Act and subsequent creation of the United States Environmental Protection Agency (USEPA) in the early 1970s, industrial, institutional and commercial entities have been required to continually improve the quality of their process wastewater effluent discharges. At the same time, population and production increases have increased water use, creating a corresponding rise in wastewater quantity. This increased water use and process wastewater generation requires more efficient removal of by-products and pollutants that allows for effluent discharge within established environmental regulatory limits. The determination of wastewater quality set forth in environmental permits has been established since the 1970s in a series of laboratory tests focused on four major categories: 1. Organics – A determination of the concentration of carbon-based (i.e., organic) compounds aimed at establishing the relative “strength” of wastewater (e.g., Biochemical Oxygen Demand (BOD), Chemical Oxygen Demand (COD), Total Organic Carbon (TOC), and Oil and Grease (O&G)). 2. Solids – A measurement of the concentration of particulate solids that can dissolve or suspend in wastewater (e.g., Total Solids (TS), Total Suspended Solids (TSS), Total Dissolved Solids (TDS), Total Volatile Solids (TVS), and Total Fixed Solids (TFS)) 3. Nutrients – A measurement of the concentration of targeted nutrients (e.g., nitrogen and phosphorus) that can contribute to the acceleration of eutrophication (i.e., the natural aging of water bodies) 4. Physical Properties and Other Impact Parameters – Analytical tests designed to measure a varied group of constituents directly impact wastewater treatability Figure 1. -

2 3 Molarity and Dilutions
Warm up • Discuss at your table: What does it mean if a solution is concentrated? What does it mean if a solution is dilute? Bonus: Try to use some new vocabulary from last week! Lab Debrief – use this data! • Class Averages: 20.4 45.2 62.9 74.6 Excel demo: graphing • Title • Units • Label axes • Intervals • Plot X: temperature Y: solubility Solubility as a function of temperature… Solutions Solvent solute When the solvent is water the solution is said to be aqueous We need a way to quantify the Concentration of Solutions Molarity (M), or molar concentration, is defined as the number of moles of solute per liter of solution. moles solute molarity = liters solution Molarity is a unit, but also can be used as a conversion factor Example 1: Molarity formula For an aqueous solution of glucose (C6H12O6), determine the molarity of a 2.00 L of a solution that contains 50.0 g of glucose Example 2: Molarity formula For an aqueous solution of glucose (C6H12O6), determine the volume of this solution that would contain 0.250 mole of glucose. Example 3: Molarity formula For an aqueous solution of glucose (C6H12O6) determine the number of moles of glucose in 0.500 L of this solution. 4.5 Example 1: Making a molar solution Explain how to make 1 liter of 0.125 M KBr solution Example 2: Making a molar solution Explain how to make 500 milliliters of 0.375 M MgCl2 solution Practice • In class: • HW: Complete lab Warm up • Discuss at your table: On a hot summer day you are sipping lemonade but it is far too sweet! What should you do if you want to dilute your beverage? Dilution is the procedure for preparing a less concentrated solution from a more concentrated solution. -

SC112 Laboratory Learning Objectives 12.5.2018 ______
AY2018-2019 SC112 Laboratory Learning Objectives 12.5.2018 _____________________________________________________________________________________ To satisfy the minimum requirements for this course, you should be able to: Experiment 20B Determination of the Solubility Of Caso4 By Ion-Exchange And By Complexometric Titration 1. Explain the principle of ion exchange. 2. Use proper experimental techniques with a column containing an ion-exchange resin. 3. Use stoichiometry to relate the data from an acid-base titration to the amount of a metal ion removed by a cation-exchange column. 4. Define a back-titration. 5. Use stoichiometry to relate the data from a complexometric titration to the amount of metal ion in a solution. Experiment 34D Evaluation of Deicer and Antifreeze Performance 1. Understand the colligative property of freezing-point depression. 2. Define the following terms: solute, solvent, solution, and molality. 3. Prepare a solution of a given molality. 4. Experimentally determine the freezing points of pure liquids and solutions. 5. Evaluate experimental data and reach conclusions based on results. Experiment 13I The Reaction of Red Food Color With Bleach 1. Relate absorbance measurements to concentrations, using the beer-lambert law. 2. Apply the method of comparing initial reaction rates to determine the order of reaction with respect to one reactant. 3. Apply the graphical (integrated rate law) method to determine the order of reaction with respect to one reactant. Experiment 12I Principles of Equilibrium 1. Select an appropriate wavelength for use in experiments involving absorbance of light. 2. Evaluate experimental data using ice tables obtain the equilibrium constant k. 3. Interpret the measurable effects of disturbances to a system at equilibrium in terms of LeChatelier’s principle.