Development of Novel PLGA Contrast Agents for Use As Ultrasound
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sulfur Hexafluoride Hazard Summary Identification
Common Name: SULFUR HEXAFLUORIDE CAS Number: 2551-62-4 RTK Substance number: 1760 DOT Number: UN 1080 Date: April 2002 ------------------------------------------------------------------------- ------------------------------------------------------------------------- HAZARD SUMMARY * Sulfur Hexafluoride can affect you when breathed in. * If you think you are experiencing any work-related health * Sulfur Hexafluoride can irritate the skin causing a rash or problems, see a doctor trained to recognize occupational burning feeling on contact. Direct skin contact can cause diseases. Take this Fact Sheet with you. frostbite. * Exposure to hazardous substances should be routinely * Sulfur Hexafluoride may cause severe eye burns leading evaluated. This may include collecting personal and area to permanent damage. air samples. You can obtain copies of sampling results * Breathing Sulfur Hexafluoride can irritate the nose and from your employer. You have a legal right to this throat. information under OSHA 1910.1020. * Breathing Sulfur Hexafluoride may irritate the lungs causing coughing and/or shortness of breath. Higher WORKPLACE EXPOSURE LIMITS exposures can cause a build-up of fluid in the lungs OSHA: The legal airborne permissible exposure limit (pulmonary edema), a medical emergency, with severe (PEL) is 1,000 ppm averaged over an 8-hour shortness of breath. workshift. * High exposure can cause headache, confusion, dizziness, suffocation, fainting, seizures and coma. NIOSH: The recommended airborne exposure limit is * Sulfur Hexafluoride may damage the liver and kidneys. 1,000 ppm averaged over a 10-hour workshift. * Repeated high exposure can cause deposits of Fluorides in the bones and teeth, a condition called “Fluorosis.” This ACGIH: The recommended airborne exposure limit is may cause pain, disability and mottling of the teeth. -

Drug-Induced Anaphylaxis in China: a 10 Year Retrospective Analysis of The
Int J Clin Pharm DOI 10.1007/s11096-017-0535-2 RESEARCH ARTICLE Drug‑induced anaphylaxis in China: a 10 year retrospective analysis of the Beijing Pharmacovigilance Database Ying Zhao1,2,3 · Shusen Sun4 · Xiaotong Li1,3 · Xiang Ma1 · Huilin Tang5 · Lulu Sun2 · Suodi Zhai1 · Tiansheng Wang1,3,6 Received: 9 May 2017 / Accepted: 19 September 2017 © The Author(s) 2017. This article is an open access publication Abstract Background Few studies on the causes of (50.1%), mucocutaneous (47.4%), and gastrointestinal symp- drug-induced anaphylaxis (DIA) in the hospital setting are toms (31.3%). A total of 249 diferent drugs were involved. available. Objective We aimed to use the Beijing Pharma- DIAs were mainly caused by antibiotics (39.3%), traditional covigilance Database (BPD) to identify the causes of DIA Chinese medicines (TCM) (11.9%), radiocontrast agents in Beijing, China. Setting Anaphylactic case reports from (11.9%), and antineoplastic agents (10.3%). Cephalospor- the BPD provided by the Beijing Center for Adverse Drug ins accounted for majority (34.5%) of antibiotic-induced Reaction Monitoring. Method DIA cases collected by the anaphylaxis, followed by fuoroquinolones (29.6%), beta- BPD from January 2004 to December 2014 were adjudi- lactam/beta-lactamase inhibitors (15.4%) and penicillins cated. Cases were analyzed for demographics, causative (7.9%). Blood products and biological agents (3.1%), and drugs and route of administration, and clinical signs and plasma substitutes (2.1%) were also important contributors outcomes. Main outcome measure Drugs implicated in DIAs to DIAs. Conclusion A variety of drug classes were impli- were identifed and the signs and symptoms of the DIA cases cated in DIAs. -

Gasket Chemical Services Guide
Gasket Chemical Services Guide Revision: GSG-100 6490 Rev.(AA) • The information contained herein is general in nature and recommendations are valid only for Victaulic compounds. • Gasket compatibility is dependent upon a number of factors. Suitability for a particular application must be determined by a competent individual familiar with system-specific conditions. • Victaulic offers no warranties, expressed or implied, of a product in any application. Contact your Victaulic sales representative to ensure the best gasket is selected for a particular service. Failure to follow these instructions could cause system failure, resulting in serious personal injury and property damage. Rating Code Key 1 Most Applications 2 Limited Applications 3 Restricted Applications (Nitrile) (EPDM) Grade E (Silicone) GRADE L GRADE T GRADE A GRADE V GRADE O GRADE M (Neoprene) GRADE M2 --- Insufficient Data (White Nitrile) GRADE CHP-2 (Epichlorohydrin) (Fluoroelastomer) (Fluoroelastomer) (Halogenated Butyl) (Hydrogenated Nitrile) Chemical GRADE ST / H Abietic Acid --- --- --- --- --- --- --- --- --- --- Acetaldehyde 2 3 3 3 3 --- --- 2 --- 3 Acetamide 1 1 1 1 2 --- --- 2 --- 3 Acetanilide 1 3 3 3 1 --- --- 2 --- 3 Acetic Acid, 30% 1 2 2 2 1 --- 2 1 2 3 Acetic Acid, 5% 1 2 2 2 1 --- 2 1 1 3 Acetic Acid, Glacial 1 3 3 3 3 --- 3 2 3 3 Acetic Acid, Hot, High Pressure 3 3 3 3 3 --- 3 3 3 3 Acetic Anhydride 2 3 3 3 2 --- 3 3 --- 3 Acetoacetic Acid 1 3 3 3 1 --- --- 2 --- 3 Acetone 1 3 3 3 3 --- 3 3 3 3 Acetone Cyanohydrin 1 3 3 3 1 --- --- 2 --- 3 Acetonitrile 1 3 3 3 1 --- --- --- --- 3 Acetophenetidine 3 2 2 2 3 --- --- --- --- 1 Acetophenone 1 3 3 3 3 --- 3 3 --- 3 Acetotoluidide 3 2 2 2 3 --- --- --- --- 1 Acetyl Acetone 1 3 3 3 3 --- 3 3 --- 3 The data and recommendations presented are based upon the best information available resulting from a combination of Victaulic's field experience, laboratory testing and recommendations supplied by prime producers of basic copolymer materials. -

BGA244 Binary Gas Analyzer User Manual
BGA244 Binary Gas Analyzer User Manual Revision 1.10 Certification Stanford Research Systems certifies that this product met its published specification at the time of shipment. Warranty This Stanford Research Systems product is warranted against defects in materials and workmanship for a period of one (1) year from the date of shipment. Service For warranty service or repair, this product must be returned to a Stanford Research Systems authorized service facility. Contact Stanford Research Systems or an authorized representative for a RMA (Return Material Authorization) Number before returning this product for repair. These are available at www.thinksrs.com under Support, Repair/Calibration. All users returning a BGA244 back to the factory for repair and/or service must submit a correctly completed “Declaration of Contamination of Equipment” form, available as part of the RMA process. The SRS personnel carrying out repair and service of the BGA244 must be informed of the condition of the components prior to any work being performed. Warning All returns to SRS must be free of harmful, corrosive, radioactive or toxic materials. Dedication In memory of Jim Williams, 1948 - 2011: Legendary Author and Analog Design Guru Information in this document is subject to change without notice. Copyright © Stanford Research Systems, Inc., 2016-2018. All rights reserved. Stanford Research Systems, Inc. 1290-C Reamwood Avenue Sunnyvale, California 94089 Phone: (408) 744-9040 Fax: (408) 744-9049 Email: [email protected] www.thinksrs.com Printed in the -

ACR Manual on Contrast Media
ACR Manual On Contrast Media 2021 ACR Committee on Drugs and Contrast Media Preface 2 ACR Manual on Contrast Media 2021 ACR Committee on Drugs and Contrast Media © Copyright 2021 American College of Radiology ISBN: 978-1-55903-012-0 TABLE OF CONTENTS Topic Page 1. Preface 1 2. Version History 2 3. Introduction 4 4. Patient Selection and Preparation Strategies Before Contrast 5 Medium Administration 5. Fasting Prior to Intravascular Contrast Media Administration 14 6. Safe Injection of Contrast Media 15 7. Extravasation of Contrast Media 18 8. Allergic-Like And Physiologic Reactions to Intravascular 22 Iodinated Contrast Media 9. Contrast Media Warming 29 10. Contrast-Associated Acute Kidney Injury and Contrast 33 Induced Acute Kidney Injury in Adults 11. Metformin 45 12. Contrast Media in Children 48 13. Gastrointestinal (GI) Contrast Media in Adults: Indications and 57 Guidelines 14. ACR–ASNR Position Statement On the Use of Gadolinium 78 Contrast Agents 15. Adverse Reactions To Gadolinium-Based Contrast Media 79 16. Nephrogenic Systemic Fibrosis (NSF) 83 17. Ultrasound Contrast Media 92 18. Treatment of Contrast Reactions 95 19. Administration of Contrast Media to Pregnant or Potentially 97 Pregnant Patients 20. Administration of Contrast Media to Women Who are Breast- 101 Feeding Table 1 – Categories Of Acute Reactions 103 Table 2 – Treatment Of Acute Reactions To Contrast Media In 105 Children Table 3 – Management Of Acute Reactions To Contrast Media In 114 Adults Table 4 – Equipment For Contrast Reaction Kits In Radiology 122 Appendix A – Contrast Media Specifications 124 PREFACE This edition of the ACR Manual on Contrast Media replaces all earlier editions. -

Chromatographic Analysis of Pharmaceuticals Second Edition, Revised and Expanded
Chromatographic Analysis of Pharmaceuticals Second Edition, Revised and Expanded edited by John A. Adamovics Cytogen Corporation Princeton, New Jersey Marcel Dekker, Inc. New York-Basel «Hong Kong Preface ISBN: 0-8247-9776-0 The first edition of Chromatographic Analysis of Pharmaceuticals was The publisher offers discounts on this book when ordered in bulk quanti published in 1990. The past years have allowed me to evaluate leads that I ties. For more information, write to Special Sales/Professional Marketing uncovered during the researching of the first edition, such as the first pub at the address below. lished example of the application of chromatography to pharmaceutical analysis of medicinal plants. This and other examples are found in a rela This book is printed on acid-free paper. tively rare book, Uber Kapillaranalyse und ihre Anwendung in Pharmazeu- tichen Laboratorium (Leipzig, 1992), by H. Platz. Capillary analysis, the Copyright © 1997 by Marcel Dekker, Inc. All Rights Reserved. chromatographic technique used, was developed by Friedlieb Runge in the mid-1850s and was later refined by Friedrich Goppelsroeder. The principle Neither this book nor any part may be reproduced or transmitted in any of the analysis was that substances were absorbed on filter paper directly form or by any means, electronic or mechanical, including photocopying, from the solutions in which they were dissolved; they then migrated to microfilming, and recording, or by any information storage and retrieval different points on the filter paper. Capillary analysis differed from paper system, without permission in writing from the publisher. chromatography in that no developing solvent was used. We find that, from these humble beginnings 150 years ago, the direct descendant of this Marcel Dekker, Inc. -

A Specialty Pharma Growth Story Disclaimer
Rovi A Specialty Pharma Growth Story Disclaimer This document has been prepared by Laboratorios Farmacéuticos Rovi, S.A. (“ROVI” or the “Company”), solely for its use during the attached presentation. The information and each of the opinions and statements contained in this document have not been verified by independent experts and, therefore, no guarantee is provided of the impartiality, accuracy, completeness or precision of the information or opinions and statements contained in this presentation. The Company and its advisors do not assume responsibility for any damage or losses that may arise from the use of this document or the information it contains. This document does not constitute an offer or invitation to acquire or subscribe shares, in accordance with the Spanish Securities Market Law of 1988 and its implementing regulations. Moreover, this document does not constitute an offer to purchase, sell or exchange securities, a solicitation of any offer to purchase, sell or exchange securities, a solicitation of any kind of voting rights, or approval in the United States of America or any other jurisdiction. Neither this document nor any part of it are of a contractual nature, and they cannot be used to form part or construe any agreement or any kind of undertaking. This presentation may contain information and statements or declarations with future projections regarding ROVI. The future projections do not constitute historical facts and are generally identifiable by the use of terms such as “expects”, “anticipates”, “believes”, “intends”, “estimates” and similar expressions. In this regard, although ROVI believes that the expectations contained in such statements are reasonable, the investors and holders of ROVI shares are advised that the information and future projections are subject to risks and uncertainties, a large part of which are difficult to foresee, and which are, in general, out of ROVI’s control. -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01 -

Guide to the Radiology Department
Guide to The Radiology Department Henal Motiwala, MD Ghada Issa, MD Jade Wong, MD ([email protected]) Pre-Test Radiology location and schedule/coverage Implications of choosing an imaging study priority Objectives Appropriate indications and contrast agents Introduction to contrast media (i.e. allergy to contrast, and contrast administration in kidney function) Introduction to MRI safety Guide to Locations Radiology Schedule Priority and coverage Ordering a Indication study Contrast Protocol agents MR safety Floor Weinberg Gudelsky Ground ER CT /Xray/US - 1st Main Radiology - LOCATIONS Breast Center NIR/VIR 2nd - Nuc Med Main reading rooms SCHEDULE & COVERAGE On-Call Contacts • Radiology In-House Home Call ED: x8-9256, #1 Resident 24/7 x 365 - Trauma: x8-8843, #1 days • Nuc Med Attending M-F 8am-5pm Evenings, Page 4198 ED/Trauma weekends • IR resident/fellow Attending Page 0015 (Do not TC official consults!) FINAL REPORTS CT & radiographs Trauma Peds CT heads ON-CALL All reports can be seen in EPIC REPORTS “PRELIM” REPORTS ED US, MR* Peds ED All STAT IP STAT: from the Latin word “Statum”, meaning ‘immediately’ • Highest priority for life threatening conditions (such as BAT, cardiovascular emergencies, intraop instrument count; any study from shock trauma and ED) CHOOSING ASAP: As soon as possible, study to be performed at the earliest STUDY opportunity, after the STAT studies Routine: performed after STAT and ASAP, within 1 shift PRIORITY *If your patient’s clinical status changes at any time and you think the study should be prioritized, please call the tech or the corresponding reading room and let us know!! • Indication: signs & symptoms • Body part(s) • Laterality when applicable ORDERING • Allergies for CT/MR • Additional history/study comments: kidney A STUDY function, dialysis status, ability to tolerate PO contrast, status post surgery etc..) • Call back number or contact on Tiger Connect (individual or team)! STUDY INDICATION • Signs, symptoms, labs (the reason for study) • “Evaluate for,” “Concern for,” “Rule out,” “s/p anything” are NOT enough. -
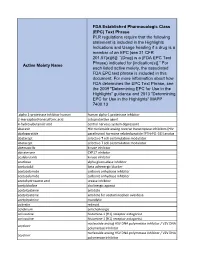
Active Moiety Name FDA Established Pharmacologic Class (EPC) Text
FDA Established Pharmacologic Class (EPC) Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for [indication(s)].” For Active Moiety Name each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. .alpha. -

Focused Ultrasound for Noninvasive, Focal Pharmacologic Neurointervention
fnins-14-00675 July 14, 2020 Time: 17:3 # 1 REVIEW published: 14 July 2020 doi: 10.3389/fnins.2020.00675 Focused Ultrasound for Noninvasive, Focal Pharmacologic Neurointervention Jeffrey B. Wang†, Tommaso Di Ianni†, Daivik B. Vyas, Zhenbo Huang, Sunmee Park, Niloufar Hosseini-Nassab, Muna Aryal and Raag D. Airan* Neuroradiology Division, Department of Radiology, Stanford University, Stanford, CA, United States A long-standing goal of translational neuroscience is the ability to noninvasively deliver therapeutic agents to specific brain regions with high spatiotemporal resolution. Focused ultrasound (FUS) is an emerging technology that can noninvasively deliver energy up the order of 1 kW/cm2 with millimeter and millisecond resolution to any point in the human brain with Food and Drug Administration-approved hardware. Although Edited by: FUS is clinically utilized primarily for focal ablation in conditions such as essential tremor, Victor Frenkel, recent breakthroughs have enabled the use of FUS for drug delivery at lower intensities University of Maryland, Baltimore, (i.e., tens of watts per square centimeter) without ablation of the tissue. In this review, we United States present strategies for image-guided FUS-mediated pharmacologic neurointerventions. Reviewed by: Vassiliy Tsytsarev, First, we discuss blood–brain barrier opening to deliver therapeutic agents of a variety University of Maryland, College Park, of sizes to the central nervous system. We then describe the use of ultrasound-sensitive United States Graeme F. Woodworth, nanoparticles to noninvasively deliver small molecules to millimeter-sized structures University of Maryland, Baltimore, including superficial cortical regions and deep gray matter regions within the brain United States without the need for blood–brain barrier opening. -

A Global Leader in Enhanced Ultrasound
1 Ultrasound From Bracco Diagnostics Inc.— A GLOBAL LEADER IN ENHANCED ULTRASOUND LUMASON FOR FOCAL LIVER LESIONS PRODUCT MONOGRAPH Our commitment to you… The ultrasound contrast agent LUMASON® (sulfur hexafluoride lipid-type A microspheres) for injectable suspension, known internationally as SonoVue®, was approved by the U.S. Food and Drug Administration (FDA) in 2014 for echocardiography, to opacify the left ventricular chamber and to improve the delineation of the left ventricular endocardial border, in adult patients with suboptimal echocardiograms. Bracco Diagnostics Inc. is proud to introduce a second indication for LUMASON. As of March 2016, LUMASON was approved for use in ultrasonography of the liver for characterization of focal liver lesions in adult and pediatric patients.1 LUMASON consists of microspheres that encapsulate an inert gas [sulfur hexafluoride (SF6)] in a phospholipid shell. LUMASON is provided in a single-use, 3-part kit that includes all components necessary for reconstitution, and requires no refrigeration for storage or mechanical agitation for reconstitution.1 Bracco Diagnostics Inc. is dedicated to providing imaging agents and solutions that improve diagnostic efficacy, patient safety, and cost effectiveness. LUMASON has been developed to meet the needs of modern ultrasound imaging practices. Please see full Prescribing Information including boxed WARNING by clicking here. 1 INDICATIONS AND USAGE LUMASON is an ultrasound contrast agent indicated for use: • in echocardiography to opacify the left ventricular