Determination of Age-Related Differences in Activation and Detoxication of Organophosphates in Rat and Human Tissues
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Environmental Properties of Chemicals Volume 2
1 t ENVIRONMENTAL 1 PROTECTION Esa Nikunen . Riitta Leinonen Birgit Kemiläinen • Arto Kultamaa Environmental properties of chemicals Volume 2 1 O O O O O O O O OO O OOOOOO Ol OIOOO FINNISH ENVIRONMENT INSTITUTE • EDITA Esa Nikunen e Riitta Leinonen Birgit Kemiläinen • Arto Kultamaa Environmental properties of chemicals Volume 2 HELSINKI 1000 OlO 00000001 00000000000000000 Th/s is a second revfsed version of Environmental Properties of Chemica/s, published by VAPK-Pub/ishing and Ministry of Environment, Environmental Protection Department as Research Report 91, 1990. The pubiication is also available as a CD ROM version: EnviChem 2.0, a PC database runniny under Windows operating systems. ISBN 951-7-2967-2 (publisher) ISBN 952-7 1-0670-0 (co-publisher) ISSN 1238-8602 Layout: Pikseri Julkaisupalvelut Cover illustration: Jussi Hirvi Edita Ltd. Helsinki 2000 Environmental properties of chemicals Volume 2 _____ _____________________________________________________ Contents . VOLUME ONE 1 Contents of the report 2 Environmental properties of chemicals 3 Abbreviations and explanations 7 3.1 Ways of exposure 7 3.2 Exposed species 7 3.3 Fffects________________________________ 7 3.4 Length of exposure 7 3.5 Odour thresholds 8 3.6 Toxicity endpoints 9 3.7 Other abbreviations 9 4 Listofexposedspecies 10 4.1 Mammais 10 4.2 Plants 13 4.3 Birds 14 4.4 Insects 17 4.5 Fishes 1$ 4.6 Mollusca 22 4.7 Crustaceans 23 4.8 Algae 24 4.9 Others 25 5 References 27 Index 1 List of chemicals in alphabetical order - 169 Index II List of chemicals in CAS-number order -

20110628140658587.Pdf
1210 YUE ET AL.:JOURNAL OF AOAC INTERNATIONAL VOL. 91, NO. 5, 2008 SPECIAL GUEST EDITOR SECTION High-Performance Thin-Layer Chromatographic Analysis of Selected Organophosphorous Pesticide Residues in Tea YONGDE YUE International Center for Bamboo and Rattan, 100102, Beijing, People’s Republic of China RONG ZHANG and WEI FAN Key Laboratory for Agri-Food Safety of Anhui Province, School of Resources and Environment, Anhui Agricultural University, Hefei, Anhui, 230036, People’s Republic of China FENG TANG International Center for Bamboo and Rattan, 100102, Beijing, People’s Republic of China The separation of 9 organophosphates determination procedure. In many cases, searching for a fast (monocrotophos, quinalphos, triazophos, and efficient method for enrichment and determination of parathion-methyl, isofenphos-methyl, temephos, organophosphorous residues from complex matrixes is still a parathion, phoxim-ethyl, and chlorpyrifos) by challenge for many researchers. high-performance thin-layer chromatography In many analytical situations, modern quantitative (HPTLC) with automated multiple development was high-performance thin-layer chromatography (HPTLC), studied. The HPTLC method was developed and when properly performed by well-trained analysts, has many validated for analysis of residues of phoxim-ethyl advantages over HPLC or GC. These advantages include and chlorpyrifos in tea. The sample was extracted simplicity of operation, the availability of many sensitive and with acetonitrile and cleaned up by ENVI-CARB selective reagents for detection and confirmation without solid-phase extraction. The extract was directly interference from the mobile phase, the ability to repeat applied as bands to glass-backed silica gel 60F254 detection and quantification at any time with changed HPTLC plates. -

Development of a Heterologous Enzyme-Linked Immunosorbent Assay for Organophosphorus Pesticides with Phage-Borne Peptide
HHS Public Access Author manuscript Author Manuscript Author ManuscriptRSC Adv Author Manuscript. Author manuscript; Author Manuscript available in PMC 2015 August 17. Published in final edited form as: RSC Adv. 2014 January 1; 4(80): 42445–42453. doi:10.1039/C4RA07059C. Development of a heterologous enzyme-linked immunosorbent assay for organophosphorus pesticides with phage-borne peptide Xiude Huaa,b, Xiaofeng Liua,b, Haiyan Shia,b, Yanru Wangc, Hee Joo Kimc, Shirley J. Geec, Minghua Wanga,b,*, Fengquan Liud,*, and Bruce D. Hammockc aCollege of Plant Protection (State & Local Joint Engineering Research Center of Green Pesticide Invention and Application), Nanjing Agricultural University, Nanjing 210095, China bKey Laboratory of Integrated Management of Crop Diseases and Pests (Nanjing Agricultural University), Ministry of Education cDepartment of Entomology and UCD Cancer Center, University of California, Davis, California 95616, United States dInstitute of Plant Protection, Jiangsu Academy of Agricultural Science, Nanjing 210014, China Abstract An enzyme-linked immunosorbent assay (ELISA) was developed to detect organophosphorus pesticides using a phage-borne peptide that was isolated from a cyclic 8-residue peptide phage −1 library. The IC50 values of the phage ELISA ranged from 1.4 to 92.1 μg L for eight organophosphorus pesticides (parathion-methyl, parathion, fenitrothion, cyanophos, EPN, paraoxon-methyl, paraoxon, fenitrooxon). The sensitivity was improved 120- and 2-fold compared to conventional homologous and heterologous ELISA, respectively. The selectivity of the phage ELISA was evaluated by measuring its cross-reactivity with 23 organophosphorus pesticides, among which eight were the main cross-reactants. The spike recoveries were between 66.1% and 101.6% for the detection of single pesticide residues of parathion-methyl, parathion and fenitrothion in Chinese cabbage, apple and greengrocery, and all of the coefficient of variation were less than or equal to 15.9%. -

Sister Chromatid Exchanges Induced by the Organophosphorus Insecticides Methyl Parathion, Dimethoate, Phoxim and Methyl Azinfos in Cultured Human Lymphocytes
Contam. Arnbient. 3. 63-70. 1981 SISTER CHROMATID EXCHANGES INDUCED BY THE ORGANOPHOSPHORUS INSECTICIDES METHYL PARATHION, DIMETHOATE, PHOXIM AND METHYL AZINFOS IN CULTURED HUMAN LYMPHOCYTES SANDRAGOMEZ-ARROYO, NORMANORIEGA-ALDANA, DOLORESJUAREZ-RODRI- GUEZ and RAFAELVILLALOBOS-PIETRINI Laboratorio de Citogenética y Mutagénesis Ambientales, Centro de Ciencias de la Atmós- fera, Universidad Nacional Autánoma de México, Coyoacán 04510, México, D.F. México (Recibido septiembre 1987, Aceptado diciembre 1987) ABSTRACT The cytogenetic effect of the organophosphorus insecticides methyl parathion or folidol. dimethoate or rogor. phoxirn or bay 77488 and methyl azinfos or gusathion was evahiated by means of the productions of sister chromatid exchanges (SCE) in cultured human lymphocytes. Positive results were obtained with methyl parathion, dimethoate and phoxim, while a negative response was achieved with methyl azinfos. The highest con- centrations of all substances decreased the SCE. possibly due to their toxicity, produ- cing the death of the damaged cells and therefore eiiminating the SCE expression. RESUMEN Se evaluó el afecto citogenbtico de los insecticidas organofosforados metil paration o foiidol, dimetoato o rogor. foxim o bay 77488 y metil azinfos o gusation mediante el anáiisis de intercambio de cromátidas hermanas (ICH) en cultivo de linfocitos humanos. Se obtuvieron resultados positivos con metil paratión, dimetoato y foxim, en tanto que con metil azinfos la respuesta fue negativa. A las concentraciones más altas de todas las sustancias empleadas disminuyeron los ICH, debido posiblemente a su elevada to- xicidad, provocando la muerte de las células más dañadas e impidiendo de esta manera la expresión del ICH. INTRODUCTION The organophosphorus insecticides appear for the first time in 1945 in Gerrnany. -
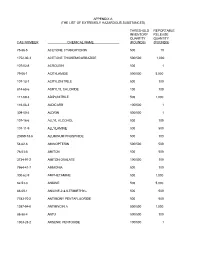
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

Organophosphate Insecticides
CHAPTER 4 HIGHLIGHTS Organophosphate Insecticides Acts through phosphorylation of the acetylcholinesterase enzyme Since the removal of organochlorine insecticides from use, organophosphate at nerve endings insecticides have become the most widely used insecticides available today. More Absorbed by inhalation, than forty of them are currently registered for use and all run the risk of acute ingestion, and skin and subacute toxicity. Organophosphates are used in agriculture, in the home, penetration in gardens, and in veterinary practice. All apparently share a common mecha- Muscarinic, nicotinic & CNS nism of cholinesterase inhibition and can cause similar symptoms. Because they effects share this mechanism, exposure to the same organophosphate by multiple routes or to multiple organophosphates by multiple routes can lead to serious additive Signs and Symptoms: toxicity. It is important to understand, however, that there is a wide range of Headache, hypersecretion, toxicity in these agents and wide variation in cutaneous absorption, making muscle twitching, nausea, specific identification and management quite important. diarrhea Respiratory depression, seizures, loss of consciousness Toxicology Miosis is often a helpful Organophosphates poison insects and mammals primarily by phosphory- diagnostic sign lation of the acetylcholinesterase enzyme (AChE) at nerve endings. The result is a loss of available AChE so that the effector organ becomes overstimulated by Treatment: the excess acetylcholine (ACh, the impulse-transmitting substance) in the nerve Clear airway, improve tissue ending. The enzyme is critical to normal control of nerve impulse transmission oxygenation from nerve fibers to smooth and skeletal muscle cells, glandular cells, and Administer atropine sulfate autonomic ganglia, as well as within the central nervous system (CNS). -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -

Decontamination of Cyanophos Residues from Water Containing Catfish (Clarias Lazera) Using Activated Charcoal and Rice Husk Ash
Journal of Applied Sciences Research 5(7): 820-826, 2009 © 2009, INSInet Publication Decontamination of Cyanophos Residues from Water Containing Catfish (Clarias Lazera) Using Activated Charcoal and Rice Husk Ash 1 Ahmed A. Romeh, 2 Mohamed B. Ashour, 2 Mohamed Y.Hendawi and 1Rady A. Ramadan 1Plant Protection Depart. Efficient Productivity. Inst., Zagazig Univ., Egypt. 2 Plant Protection Dept. Faculty of Agriculture. Zagazig Univ. Egypt. Abstract: The effect of activated charcoal and rice husk ash addition on the residues of cyanophos in water and some organs of catfish (clarias lazera) and their effect on some biochemical changes in blood serum were studies under laboratory conditions. The obtained results indicated that cyanophos showed high degradation in aquaria water compared with tap water. This was pronounced with the all considered post- treatment intervals. The average concentration of cyanophos in tap water and in water containing fish 2hrs after treatment was 8.73 and 6.60 Ug/ml respectively. These amounts were significantly decreased by time till reached 4.13 and 1.20 Ug/ml after 144hrs, representing 55.73and 87.14% dislodges of the initial amounts, respectively. Penetrated amount of cyanophos significantly increased and accumulated in the inner organs as time lapsed to reach the maximum level in the gills after 24 hrs (17.39 ug/gm) and in the liver during 48 hrs (24.51 ug/gm) while in muscles during 96 hrs (7.98 ug/gm) of experimental span. Thereafter, cyanophos began to disappear from the gills, liver and muscles gradually till the end of the experimental period (144 hrs).Addition of activated charcoal and rice husk ash to water contaminated with anilophos reduced clearly levels of residues in water and in fish gills, liver and muscles. -

Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 Theinternational Programme on Chemical Safety (IPCS) Was Established in 1980
The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 cation Hazard of Pesticides by and Guidelines to Classi The WHO Recommended Classi The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 TheInternational Programme on Chemical Safety (IPCS) was established in 1980. The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. This publication was developed in the IOMC context. The contents do not necessarily reflect the views or stated policies of individual IOMC Participating Organizations. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase international coordination in the field of chemical safety. The Participating Organizations are: FAO, ILO, UNDP, UNEP, UNIDO, UNITAR, WHO, World Bank and OECD. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment. WHO recommended classification of pesticides by hazard and guidelines to classification, 2019 edition ISBN 978-92-4-000566-2 (electronic version) ISBN 978-92-4-000567-9 (print version) ISSN 1684-1042 © World Health Organization 2020 Some rights reserved. -

Florida Pesticide Reporting Guidelines
Florida Pesticide Reporting guidelines This list is compiled from the Environmental Protection Agency List of Lists (2015) and updated with common Pesticide Trade Names. This is meant as a supplement to the EPA list of lists to clarify and assist handlers and responders in the field to Florida reporting requirements and the more common chemical nomenclature. Threshold Planning Quantity (TPQ) – The presence of Extremely Hazardous Substances (EHSs) in quantities at or above the Threshold Planning Quantity (TPQ) requires certain emergency planning activities to be conducted. The consolidated list presents the TPQ (in pounds) for section 302 chemicals in the column following the CAS number. For chemicals that are solids, there are two TPQs given (e.g., 500/10,000). In these cases, the lower quantity applies for solids in powder form with particle size less than 100 microns, or if the substance is in solution or in molten form. Otherwise, the 10,000 pound TPQ applies. Section 304 RQ‐ Facilities must immediately report accidental releases of EHS chemicals and "hazardous substances" in quantities greater than corresponding Reportable Quantities (RQs) defined under the Comprehensive Environmental Response, Compensation, and Liability Act (CERCLA) to state and local officials. Information about accidental chemical releases must be available to the public, Florida Reporting requirements below. CERCLA RQ‐ Releases of CERCLA hazardous substances, in quantities equal to or greater than their reportable quantity (RQ) in pounds, are subject to reporting to the Florida Reporting requirements below. Florida Reporting requirements: National Response Center Florida State Watch Office (800) 424-8802 (800) 320-0519 or (850) 815-4001 Florida Department of Environmental Protection Spill reporting requirements https://floridadep.gov/pollutionnotice Florida Division of Emergency Management 2555 Shumard Oak blvd. -

United States Patent (19) 11 Patent Number: 5,703,064 Yokoi Et Al
US005703064A United States Patent (19) 11 Patent Number: 5,703,064 Yokoi et al. 45) Date of Patent: Dec. 30, 1997 54 PESTICIDAL COMBINATIONS FOREIGN PATENT DOCUMENTS 75 Inventors: Shinji Yokoi; Akira Nishida, both of 0 196524 10/1986 European Pat. Of.. Shiga-ken; Tokio Obata; Kouichi Golka, both of Ube, all of Japan OTHER PUBLICATIONS 73) Assignees: Sankyo Company, Limited, Tokyo; Worthing et al, The Pesticide Manual, 9th Ed. (1991), pp. Ube industries Ltd., Ube, both of 747 and 748. Japan L.C. Gaughan et al., "Pesticide interactions: effects of orga nophosphorus pesticides on the metabolism, toxicity, and 21 Appl. No.: 405,795 persistence of selected pyrethroid insecticides". Chemical Abstracts, vol. 94, No. 9, 1981, No. 59740k of Pestic. 22 Filed: Mar 16, 1995 Biochem. Physio., vol. 14, No. 1, 1980, pp. 81-85. 30 Foreign Application Priority Data I. Ishaaya et al., "Cypermethrin synergism by pyrethroid esterase inhibitors in adults of the whitefly Bemisia tabaci". Mar 16, 1994 JP Japan ............................ HE6045.405 Chemical Abstracts, vol. 107, No. 9, 1987, No. 72818y of (51) Int. Cl................. A01N 43/54; A01N 57/00 Pestic Biochem. Physiol., vol. 28, No. 2, 1987, pp. 155-162. 52 U.S. C. ......................................... 51480; 514/256 (58) Field of Search ..................................... 514/80, 256 Primary Examiner Allen J. Robinson Attorney, Agent, or Firm-Frishauf, Holtz, Goodman, 56 References Cited Langer & Chick, P.C. U.S. PATENT DOCUMENTS 57 ABSTRACT 4,374,833 2/1983 Badmin et al. ...................... 424/225 Combinations of the known compound pyrimidifen with 4,845,097 7/1989 Matsumoto et al... 514/234.2 phosphorus-containing pesticides have a synergistic pesti 4,935,516 6/1990 Ataka et al. -

Free of Analytes Matrixes
FREE OF ANALYTES MATRIXES Matrix: Green beans Description: Green beans in plastic container sealed with pressure lid and screw cap. Approximated weight: 50g Date of analysis: May 2019 Analyzed analytes: Pesticides residues (See annex). Results: There was no presence of any of the target analytes (See annex) above the quantification limit. NOTES The material has been kept in temperature-controlled freezer and is sent frozen. Upon receive store in freezer until analysis. The test material has been analyzed by the TestQual’s collaborator laboratory accredited for the analysis of the annexed pesticides, therefore ensuring the lack of pesticides in concentrations higher than the limit of quantification of the laboratory: 10µg/Kg The list of the analyzed pesticides is the one TestQual works with regularly in our proficiency test of pesticides residues. You can check the full list of analytes in the protocol or request the documentation using the contact data on our website: http://testqual.com/contacto/?lng=en Page 1 / 3 ANNEX 2-Phenylphenol Carbendazim Dichlormid Fenazaquin 3,5-Dichloroaniline Carbophenothion Dichlobenil Fenbuconazole 3-Hydroxy-carbofuran Carbofuran Diclobutrazol Fenbutatin oxide 4,4-Dichlorobenzophenone Chlorantraniliprole Dichlofluanid Fenchlorphos Abamectin Chlorbromuron Diclofop-methyl Fenhexamid Acephate Chlorfenapyr Dicloran Fenitrothion Acetamiprid Chlorfenvinphos Dicrotophos Fenoxycarb Acetochlor Chlormephos Dieldrin Fenpropathrin Aclonifen Chloroneb Diethofencarb Fenpropimorph Acrinathrin Chloropropylate Difenoconazole