RNA-Seq As a Tool for Evaluating Human Embryo Competence
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CD56+ T-Cells in Relation to Cytomegalovirus in Healthy Subjects and Kidney Transplant Patients
CD56+ T-cells in Relation to Cytomegalovirus in Healthy Subjects and Kidney Transplant Patients Institute of Infection and Global Health Department of Clinical Infection, Microbiology and Immunology Thesis submitted in accordance with the requirements of the University of Liverpool for the degree of Doctor in Philosophy by Mazen Mohammed Almehmadi December 2014 - 1 - Abstract Human T cells expressing CD56 are capable of tumour cell lysis following activation with interleukin-2 but their role in viral immunity has been less well studied. The work described in this thesis aimed to investigate CD56+ T-cells in relation to cytomegalovirus infection in healthy subjects and kidney transplant patients (KTPs). Proportions of CD56+ T cells were found to be highly significantly increased in healthy cytomegalovirus-seropositive (CMV+) compared to cytomegalovirus-seronegative (CMV-) subjects (8.38% ± 0.33 versus 3.29%± 0.33; P < 0.0001). In donor CMV-/recipient CMV- (D-/R-)- KTPs levels of CD56+ T cells were 1.9% ±0.35 versus 5.42% ±1.01 in D+/R- patients and 5.11% ±0.69 in R+ patients (P 0.0247 and < 0.0001 respectively). CD56+ T cells in both healthy CMV+ subjects and KTPs expressed markers of effector memory- RA T-cells (TEMRA) while in healthy CMV- subjects and D-/R- KTPs the phenotype was predominantly that of naïve T-cells. Other surface markers, CD8, CD4, CD58, CD57, CD94 and NKG2C were expressed by a significantly higher proportion of CD56+ T-cells in healthy CMV+ than CMV- subjects. Functional studies showed levels of pro-inflammatory cytokines IFN-γ and TNF-α, as well as granzyme B and CD107a were significantly higher in CD56+ T-cells from CMV+ than CMV- subjects following stimulation with CMV antigens. -

Euchromatin Histone Methyltransferase II (EHMT2) Regulates the Expression of Ras-Related GTP Binding C (RRAGC) Protein
BMB Rep. 2020; 53(11): 576-581 BMB www.bmbreports.org Reports Euchromatin histone methyltransferase II (EHMT2) regulates the expression of ras-related GTP binding C (RRAGC) protein Supyong Hwang1, Soyoung Kim1, Kyungkon Kim1,2,3, Jeonghun Yeom2, Sojung Park1 & Inki Kim1,2,3,* 1Biomedical Research Center, ASAN Institute for Life Sciences, ASAN Medical Center, Seoul 05505, 2Convergence Medicine Research Center (CREDIT), ASAN Institute for Life Sciences, ASAN Medical Center, Seoul 05505, 3Department of Convergence Medicine, University of Ulsan College of Medicine, Seoul 05505, Korea Dimethylation of the histone H3 protein at lysine residue 9 INTRODUCTION (H3K9) is mediated by euchromatin histone methyltransferase II (EHMT2) and results in transcriptional repression of target Epigenetic modifications are gene regulatory mechanisms that genes. Recently, chemical inhibition of EHMT2 was shown to are independent of changes in DNA sequences (1). This mode induce various physiological outcomes, including endoplasmic of gene regulation can be achieved by means of histone and reticulum stress-associated genes transcription in cancer cells. DNA modifications, such as methylation and acetylation (1). To identify genes that are transcriptionally repressed by EHMT2 Among these, methylation at lysine residues 4 (H3K4) and 36 during apoptosis, and cell stress responses, we screened genes (H3K36) of histone H3 are hallmarks of transcriptional acti- that are upregulated by BIX-01294, a chemical inhibitor of vation, whereas methylation of histone H3 residues at lysines EHMT2. RNA sequencing analyses revealed 77 genes that were 9 (H3K9) and 27 (H3K27) leads to repression (2). Methylation upregulated by BIX-01294 in all four hepatic cell carcinoma of histones is accomplished by histone methyltransferases (HMTs) (HCC) cell lines. -

Effects of Rapamycin on Social Interaction Deficits and Gene
Kotajima-Murakami et al. Molecular Brain (2019) 12:3 https://doi.org/10.1186/s13041-018-0423-2 RESEARCH Open Access Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero Hiroko Kotajima-Murakami1,2, Toshiyuki Kobayashi3, Hirofumi Kashii1,4, Atsushi Sato1,5, Yoko Hagino1, Miho Tanaka1,6, Yasumasa Nishito7, Yukio Takamatsu7, Shigeo Uchino1,2 and Kazutaka Ikeda1* Abstract The mammalian target of rapamycin (mTOR) signaling pathway plays a crucial role in cell metabolism, growth, and proliferation. The overactivation of mTOR has been implicated in the pathogenesis of syndromic autism spectrum disorder (ASD), such as tuberous sclerosis complex (TSC). Treatment with the mTOR inhibitor rapamycin improved social interaction deficits in mouse models of TSC. Prenatal exposure to valproic acid (VPA) increases the incidence of ASD. Rodent pups that are exposed to VPA in utero have been used as an animal model of ASD. Activation of the mTOR signaling pathway was recently observed in rodents that were exposed to VPA in utero, and rapamycin ameliorated social interaction deficits. The present study investigated the effect of rapamycin on social interaction deficits in both adolescence and adulthood, and gene expressions in mice that were exposed to VPA in utero. We subcutaneously injected 600 mg/kg VPA in pregnant mice on gestational day 12.5 and used the pups as a model of ASD. The pups were intraperitoneally injected with rapamycin or an equal volume of vehicle once daily for 2 consecutive days. The social interaction test was conducted in the offspring after the last rapamycin administration at 5–6 weeks of ages (adolescence) or 10–11 weeks of age (adulthood). -

Regulation of Hematopoietic Activity Involving New Interacting Partners (RRAGC & PSMC2, CKAP4 & MANF and CTR9 & CNTNAP2)
CellBio, 2020, 9, 123-141 https://www.scirp.org/journal/cellbio ISSN Online: 2325-7792 ISSN Print: 2325-7776 Regulation of Hematopoietic Activity Involving New Interacting Partners (RRAGC & PSMC2, CKAP4 & MANF and CTR9 & CNTNAP2) Swati Sharma1, Gurudutta U. Gangenahalli2*, Upma Singh3 1Department of Pharmacology, All India Institute of Medical Science (AIIMS), New Delhi, India 2Stem Cell & Gene Therapy Research Group, Institute of Nuclear Medicine & Allied Sciences, New Delhi, India 3Department of Applied Chemistry, School of Vocational Studies & Applied Sciences, Gautam Buddha University, Greater Noida, India How to cite this paper: Sharma, S., Gan- Abstract genahalli, G.U. and Singh, U. (2020) Regu- lation of Hematopoietic Activity Involving Hematopoietic stem cells (HSCs) are tissue-specific cells giving rise to all New Interacting Partners (RRAGC & PSMC2, mature blood cell types regulated by a diverse group of hematopoietic cyto- CKAP4 & MANF and CTR9 & CNTNAP2). kines and growth factors that influences the survival & proliferation of early CellBio, 9, 123-141. https://doi.org/10.4236/cellbio.2020.93007 progenitors and differentiation mechanisms by modulating the functional ac- tivities of HSCs. In this study, the functional yet distinctive role of three novel Received: August 13, 2020 combinations of gene pairs RRAGC & PSMC2; CKAP4 & MANF; and CTR9 Accepted: September 27, 2020 & CNTNAP2 have been newly identified. These novel combinations of genes Published: September 30, 2020 were confirmed and expressed in K562 human leukemic cell line in the pres- Copyright © 2020 by author(s) and ence of cytokine combination (IL-3, FLT-3 and SCF) using RT-PCR and Scientific Research Publishing Inc. siRNA-mediated gene knock down strategy. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -
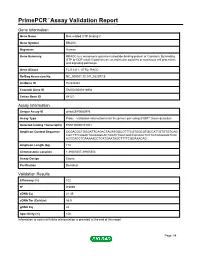
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Ras-related GTP binding C Gene Symbol RRAGC Organism Human Gene Summary RRAGC is a monomeric guanine nucleotide-binding protein or G protein. By binding GTP or GDP small G proteins act as molecular switches in numerous cell processes and signaling pathways. Gene Aliases FLJ13311, GTR2, RAGC RefSeq Accession No. NC_000001.10, NT_032977.9 UniGene ID Hs.532461 Ensembl Gene ID ENSG00000116954 Entrez Gene ID 64121 Assay Information Unique Assay ID qHsaCEP0050976 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000373001 Amplicon Context Sequence CCGACGCTGGGATTCAGACTAGATGGCGTTTCGTGGCGTGCCATTGTGTGTCAG CGCTTTCAGACTGGAGGCACTAGTCTGGTGACCACAGCTCCTGTGAGAAGTCAC ACCCACCTCAAAAACCTCATGAATAGCTTTTCGGAAACAG Amplicon Length (bp) 118 Chromosome Location 1:39305207-39305354 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 102 R2 0.9989 cDNA Cq 21.35 cDNA Tm (Celsius) 85.5 gDNA Cq 24 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report RRAGC, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Recurrent Mutations in the MTOR Regulator RRAGC in Follicular Lymphoma Zhang Xiao Ying1, Meiyan Jin2, Luke F
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0609 Biology of Human Tumors Clinical Cancer Research Recurrent Mutations in the MTOR Regulator RRAGC in Follicular Lymphoma Zhang Xiao Ying1, Meiyan Jin2, Luke F. Peterson1, Denzil Bernard1, Kamlai Saiya-Cork1, Mehmet Yildiz1, Shaomeng Wang1, Mark S. Kaminski1, Alfred E. Chang3, Daniel J. Klionsky2, and Sami N. Malek1 Abstract Purpose: This study was performed to further our understand- Mutations in RRAGC distinctly clustered on one protein surface ing of the biological and genetic basis of follicular lymphoma and area surrounding the GTP/GDP–binding sites. Mutated RRAGC to identify potential novel therapy targets. proteins demonstrated increased binding to RPTOR (raptor) and Experimental Design: We analyzed previously generated substantially decreased interactions with the product of the tumor whole exome sequencing data of 23 follicular lymphoma cases suppressor gene FLCN (folliculin). In stable retrovirally trans- and one transformed follicular lymphoma case and expanded fected 293T cells, cultured in the presence or absence of leucine, findings to a combined total of 125 follicular lymphoma/3 multiple RRAGC mutations demonstrated elevated MTOR acti- transformed follicular lymphoma. We modeled the three- vation as evidenced by increased RPS6KB/S6-kinase phosphory- dimensional location of RRAGC-associated hotspot muta- lation. Similar activation phenotypes were uncovered in yeast tions. We performed functional studies on novel RRAGC engineered to express mutations in -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Agricultural University of Athens
ΓΕΩΠΟΝΙΚΟ ΠΑΝΕΠΙΣΤΗΜΙΟ ΑΘΗΝΩΝ ΣΧΟΛΗ ΕΠΙΣΤΗΜΩΝ ΤΩΝ ΖΩΩΝ ΤΜΗΜΑ ΕΠΙΣΤΗΜΗΣ ΖΩΙΚΗΣ ΠΑΡΑΓΩΓΗΣ ΕΡΓΑΣΤΗΡΙΟ ΓΕΝΙΚΗΣ ΚΑΙ ΕΙΔΙΚΗΣ ΖΩΟΤΕΧΝΙΑΣ ΔΙΔΑΚΤΟΡΙΚΗ ΔΙΑΤΡΙΒΗ Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων ΕΙΡΗΝΗ Κ. ΤΑΡΣΑΝΗ ΕΠΙΒΛΕΠΩΝ ΚΑΘΗΓΗΤΗΣ: ΑΝΤΩΝΙΟΣ ΚΟΜΙΝΑΚΗΣ ΑΘΗΝΑ 2020 ΔΙΔΑΚΤΟΡΙΚΗ ΔΙΑΤΡΙΒΗ Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων Genome-wide association analysis and gene network analysis for (re)production traits in commercial broilers ΕΙΡΗΝΗ Κ. ΤΑΡΣΑΝΗ ΕΠΙΒΛΕΠΩΝ ΚΑΘΗΓΗΤΗΣ: ΑΝΤΩΝΙΟΣ ΚΟΜΙΝΑΚΗΣ Τριμελής Επιτροπή: Aντώνιος Κομινάκης (Αν. Καθ. ΓΠΑ) Ανδρέας Κράνης (Eρευν. B, Παν. Εδιμβούργου) Αριάδνη Χάγερ (Επ. Καθ. ΓΠΑ) Επταμελής εξεταστική επιτροπή: Aντώνιος Κομινάκης (Αν. Καθ. ΓΠΑ) Ανδρέας Κράνης (Eρευν. B, Παν. Εδιμβούργου) Αριάδνη Χάγερ (Επ. Καθ. ΓΠΑ) Πηνελόπη Μπεμπέλη (Καθ. ΓΠΑ) Δημήτριος Βλαχάκης (Επ. Καθ. ΓΠΑ) Ευάγγελος Ζωίδης (Επ.Καθ. ΓΠΑ) Γεώργιος Θεοδώρου (Επ.Καθ. ΓΠΑ) 2 Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων Περίληψη Σκοπός της παρούσας διδακτορικής διατριβής ήταν ο εντοπισμός γενετικών δεικτών και υποψηφίων γονιδίων που εμπλέκονται στο γενετικό έλεγχο δύο τυπικών πολυγονιδιακών ιδιοτήτων σε κρεοπαραγωγικά ορνίθια. Μία ιδιότητα σχετίζεται με την ανάπτυξη (σωματικό βάρος στις 35 ημέρες, ΣΒ) και η άλλη με την αναπαραγωγική -

Dasatinib Is an Effective Treatment for Angioimmunoblastic T-Cell
Supplemental information Dasatinib Is An Effective Treatment For Angioimmunoblastic T-Cell Lymphoma Tran B. Nguyen1+, Mamiko Sakata-Yanagimoto1,2+*, Manabu Fujisawa3, Sharna Tanzima Nuhat3, Hiroaki Miyoshi4, Yasuhito Nannya5, Koichi Hashimoto6, Kota Fukumoto3, Olivier A. Bernard7, Yusuke Kiyoki2, Kantaro Ishitsuka2, Haruka Momose2, Shinichiro Sukegawa2, Atsushi Shinagawa8, Takuya Suyama8, Yuji Sato9, Hidekazu Nishikii1,2, Naoshi Obara1,2, Manabu Kusakabe1,2, Shintaro Yanagimoto10, Seishi Ogawa5, Koichi Ohshima4, and Shigeru Chiba1,2,11* 1. Department of Hematology, Faculty of Medicine, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8575, Japan. 2. Department of Hematology, University of Tsukuba Hospital, 2-1-1 Amakubo, Tsukuba, Ibaraki 305-8576, Japan. 3. Department of Hematology, Graduate School of Comprehensive Human Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8575, Japan. 4. Department of Pathology, Kurume University, School of Medicine, 67 Asahi, Kurume, Fukuoka 830-0011, Japan. 5. Department of Pathology and Tumor Biology, Graduate School of Medicine, Kyoto University, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan. 6. Tsukuba Clinical Research and Development Organization (TCReDo), University of Tsukuba, 1- 1-1 Tennodai, Tsukuba, Ibaraki 305-8575, Japan. 7. INSERM U1170, Gustave Roussy, Université Paris-Saclay, Equipe Labellisée Ligue Nationale Contre le Cancer, Villejuif, France. 8. Department of Hematology, Hitachi General Hospital, 2-1-1 Jonan-cho, Hitachi, Ibaraki 317-0077, Japan. 9. Department of Hematology and Oncology, Tsukuba Memorial Hospital, 1187-299 Kaname, Tsukuba, Ibaraki 300-2622, Japan. 10. Division for Health Service Promotion, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113- 0033, Japan. 11. Life Science Center for Survival Dynamics, Tsukuba Advanced Research Alliance, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8575, Japan. -
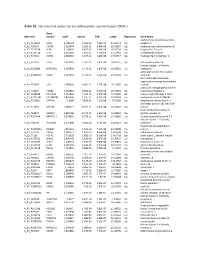
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

A Novel Rapamycin Analog Is Highly Selective for Mtorc1 in Vivo
ARTICLE https://doi.org/10.1038/s41467-019-11174-0 OPEN A novel rapamycin analog is highly selective for mTORC1 in vivo Katherine H. Schreiber1,11, Sebastian I. Arriola Apelo2,3,4,11, Deyang Yu3,4,5, Jacqueline A. Brinkman3,4, Michael C. Velarde6,9, Faizan A. Syed3,4, Chen-Yu Liao 1, Emma L. Baar3,4, Kathryn A. Carbajal3,4, Dawn S. Sherman3,4, Denise Ortiz1,7, Regina Brunauer 1,10, Shany E. Yang3,4, Stelios T. Tzannis6, Brian K. Kennedy1 & Dudley W. Lamming 3,4,5,8 1234567890():,; Rapamycin, an inhibitor of mechanistic Target Of Rapamycin Complex 1 (mTORC1), extends lifespan and shows strong potential for the treatment of age-related diseases. However, rapamycin exerts metabolic and immunological side effects mediated by off-target inhibition of a second mTOR-containing complex, mTOR complex 2. Here, we report the identification of DL001, a FKBP12-dependent rapamycin analog 40x more selective for mTORC1 than rapamycin. DL001 inhibits mTORC1 in cell culture lines and in vivo in C57BL/6J mice, in which DL001 inhibits mTORC1 signaling without impairing glucose homeostasis and with substantially reduced or no side effects on lipid metabolism and the immune system. In cells, DL001 efficiently represses elevated mTORC1 activity and restores normal gene expression to cells lacking a functional tuberous sclerosis complex. Our results demonstrate that highly selective pharmacological inhibition of mTORC1 can be achieved in vivo, and that selective inhibition of mTORC1 significantly reduces the side effects associated with conventional rapalogs. 1 Buck Institute for Research on Aging, Novato, CA 94945, USA. 2 Department of Dairy Science, University of Wisconsin-Madison, Madison, WI 53706, USA.