Development of a Novel 3D Nanofibre Co-Culture Model for Characterisation of Neural Cell Degeneration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1959 Surname
Surname Given Age Date Page Maiden Note Abercrombie James W. 36 10-Jul B-3 Abercrombie Josephine 82 27-Nov B-3 Abraham Clarence F. 64 17-Jul B-3 Absher Donna 87 18-Dec A-14 Adam Louis S. 66 21-Jan B-3 Adamczyk Klemenz 86 6-Mar B-8 Adams Elizabeth 83 10-Dec B-3 Adams Elmer L. 19 14-May A-1 Adams Myrtle M. 78 9-Mar 11 Adams Richard Dale Indant 26-Feb B-3 Adams Robert D. 62 14-Jul A-11 Ader Jeanette L. 27 29-Mar B-3 Adler Vickie Lynn 17 months 1-Jul B-3 Adley Pearl Katherine 79 9-Jul A-11 Adrien Andrew 66 29-Nov B-3 Ahlborn Bertha M. 77 8-Oct B-3 Ahlgren Oscar F. 69 23-Apr B-3 Aistgem Dawn Marie Infant 25-May A-13 Albrecht Chester 61 12-Feb B-3 Aldridge William C. (Blackie) 72 23-Mar 11 Alexander Helen H. 54 29-Sep A-11 Alford Howard E. 63 19-Apr B-3 Alger L. Nora 91 18-Jun B-3 Allen Charles J. 52 11-Mar B-3 Allison Daisy 71 21-May B-3 Allshouse Marcella Wickersham 6-Nov B-3 Ally Susan Jane 6 29-May A-1 Amanta Sister Mary 58 28-Jun B-4 Ambler Infant Son Infant 30-Aug B-3 Amey William A. 81 19-Jan 11 Amhurst Pearl J. 48 8-Apr B-3 Andersen Cordelia 79 6-Dec B-3 Anderson Bertie 72 24-May B-3 Anderson Byron 68 27-Feb B-3 Anderson Lily 68 10-Nov A-11 Anderson Roy G. -

Special Report
September 2014 turnarounds & Workouts 7 Special Report European Restructuring Practices of Major U.S. Law Firms, page 1 Firm Senior Professionals Representative Clients Bingham McCutchen James Roome Elisabeth Baltay Creditors of: Arcapita Bank, Bulgaria Telecommunications/Vivacom, Crest +44.20.7661.5300 Barry G. Russell Liz Osborne Nicholson, Dannemora Minerals, DEPFA Bank, Findus Foods, Gala Coral, www.bingham.com James Terry Neil Devaney Icelandic Banks (Kaupthing, Glitnir and Landsbanki), Invitel, Klöckner Stephen Peppiatt Emma Simmonds Pentaplast, Media Works, Northland Resources, Oceanografia, OSX3 Leasing Tom Bannister B.V., Petromena, Petroplus, Preem, Punch Taverns, Royal Imtech, Selecta, Sevan Marine, Skeie Drilling, Straumur, Technicolor S.A. (Thomson S.A.), Terreal, The Quinn Group, Uralita, Wind Hellas, Xcite, and others. Cadwalader, Wickersham Gregory Petrick Louisa Watt Centerbridge Partners, Avenue Capital Group, GSO Capital Partners, & Taft Richard Nevins Paul Dunbar Oaktree Capital Management, Varde Partners, Golden Tree Asset +44 (0) 20 7170 8700 Yushan Ng Karen McMaster Management, Bluebay Asset Management, MBIA, Davidson Kempner, www.cadwalader.com Holly Neavill Alexis Kay Outrider Management, GLG Partners, Warwick Capital, Alchemy, Finnisterre Capital. Davis Polk Donald S. Bernstein Timothy Graulich Lehman Brothers International (Europe) and its U.K. Lehman affiliates, +44 20 7418 1300 Karen E. Wagner Elliot Moskowitz Sterling Equities in Madoff SIPA liquidation, Technicolor S.A., Royal www.davispolk.com Andrés V. Gil Thomas J. Reid Imtech, Carrefour, major global banks and financial institutions in Arnaud Pérès Christophe Perchet connection with several monoline insurance company restructurings, Marshall S. Huebner John Banes Goldman Sachs in connection with exposures to BP, Castle HoldCo 4, Benjamin S. Kaminetzky Reuven B. -
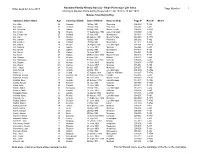
Ships Passenger List Index As of 22 June 2013
Nanaimo Family History Society - Ships Passenger List Index Index as at 22 June 2013 Page Number: 1 Arriving at Quebec Ports during the period 31 Jul 1903 to 13 Oct 1910 Names from Kot to Krg Surname, Given Name Age Country of Birth Date of Arrival Name of Ship Page # Reel # Notes Kot, Afka 26 German 16 May,1904 Phoenicia 036-036 T-482 Kot, Andre 30 German 16 May,1904 Phoenicia 036-036 T-482 Kot, Antonina 21 Galicia 05 May,1909 Mount Temple 006-005 T-4760 Kot, Chaim 42 Russia 11 September,1903 Lake Champlain 009-009 T-482 Kot, Gerard Jan 27 Holland 17 May,1905 Montezuma 013-013 T-484 Kot, Jan 35 Russia 29 August,1910 Mount Temple 003-003 T-4771 Kot, Johann 1 German 16 May,1904 Phoenicia 036-036 T-482 Kot, Lambert 22 Holland 17 May,1905 Montezuma 013-013 T-484 Kot, Marcin 28 Russia 14 August,1910 Lake Michigan 001-001 T-4770 Kot, Marscia 26 Galicia 16 June,1907 Montreal 046-044 T-490 Kot, Michal 24 Galicia 23 May,1907 Montezuma 018-018 T-489 Kot, Michel 31 Galicia 16 June,1907 Montreal 046-044 T-490 Kot, Nicol 27 Galicia 09 November, 1904 Mount Temple 010-010 T-484 Kot, Shmool 38 Russia 07 May,1905 Ottawa 003-002 T-484 Kot, Stanislaus 22 German 02 November,1907 Victorian 006-006 T-492 Kot, Stepan 23 Russia 11 June,1910 Montreal 004-003 T-4767 Kot, Wasyl 6m Galicia 16 June,1907 Montreal 046-044 T-490 Kot?, Jakub 31 Russia 03 July,1907 Montrose 007-007 T-490 Kota, Nicolai 27 Austria 23 May,1910 Lake Michigan 009-009 T-4766 Kota???, ? 21 Ireland 05 August,1909 Empress of Britain 030-007 T-4762 Kotacliko, Nicolai 26 Russian Lith 03 November,1906 -

Anuncio 709 Del BOE Núm. 6 De 2011
BOLETÍN OFICIAL DEL ESTADO Núm. 6 Viernes 7 de enero de 2011 Sec. V-B. Pág. 1429 V. Anuncios B. Otros anuncios oficiales MINISTERIO DE ECONOMÍA Y HACIENDA 709 Resolución de 29 de diciembre de 2010, del Servicio de Gestión Económica de la Agencia Estatal de Administración Tributaria, por la que se anuncian las notificaciones por comparecencia, después de haber realizado primero y segundo intentos. En virtud de lo dispuesto en el artículo 112 de la Ley General Tributaria (Ley 58/2003, de 17 de diciembre), para las notificaciones de naturaleza Tributaria, y artículo 59 de la Ley 30/1992 de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, de 26 de noviembre, en relación con las de naturaleza no Tributaria, no siendo posible practicar la notificación por causas no imputables a la Administración y habiéndose realizado, al menos, los intentos de notificación exigidos por los citados artículos, por el presente anuncio se cita a los obligados tributarios, demás interesados o representantes que se relacionan en el anexo que se publica en los suplementos de este diario, para ser notificados por comparecencia de los actos administrativos derivados de los procedimientos que en el mismo se incluyen. Los obligados tributarios, los demás interesados o sus representantes deberán comparecer en el plazo máximo de 15 días naturales, contados desde el día siguiente al de publicación del presente anuncio en el Boletín Oficial del Estado, en horario de nueve a catorce horas, de lunes a viernes, en los lugares que en cada caso se señalan, al efecto de practicarse las notificaciones pendientes en los procedimientos tramitados por los órganos relacionados en el anexo que se publica en los suplementos de este diario. -

Surname Given Age Date Page Maiden Note Aageberg Alice F. 101 11-Feb D-1 See Also Article Feb. 12, P. C-5 Abegg Frances J. 64 9
Surname Given Age Date Page Maiden Note Aageberg Alice F. 101 11-Feb D-1 See also article Feb. 12, p. C-5 Abegg Frances J. 64 9-Jun D-1 Abel Delores Ruth 57 9-Sep B-6 Abel Ralph 31-Dec D-5 Abell Thelma 80 18-Dec A-11 See article, p. A-11 Abney Raymond 83 15-Dec D-1 See article, p. D-1 Acheson Helen G. 61 26-Apr E-2 Adamczyk Richard J. 48 5-Nov C-10 Adamovich Michael 77 10-Feb D-1 Adams Altha B. 91 12-Jan C-2 Adams Corinne, Deputy 79 12-Mar C-3 See also article March 13, p. C-1 Adams Frances D. 53 10-Nov D-5 Malloy Adams Leonard Eugene 58 2-Jun D-3 (Preacher) Adams Marie Mae 75 10-Mar C-1 Adams Robert F. 72 11-Jan D-1 Adams Susan L. 35 3-Mar C-6 Adams Thomas A. 12 Februray 2 A-6 Adams Thomas P. 38 19-Jan A-8 Addison James Grover "J. G." 69 3-Apr C-1 Adelsperger Edward H. 69 25-Mar D-1 Adelsperger Helen 71 24-Nov B-5 Adkins Kathleen 70 3-Feb B-7 Adler Sophie 74 4-Feb D-1 Adolph Bernard 60 2-Oct C-2 Adzima Rose 26-May D-5 Kubeck Agee Hardy 66 13-Jan A-1 See article, p. A-1 Agerter Tim D. 22 19-May D-1 See also article, p. A-2 Ahearn Mary 82 22-Apr C-3 Doolin Akhtar Julie Wayman 26 15-Apr C-5 Alaimo Bartole (Bart) 66 24-Oct B-2 Alaimo Damiana 86 20-Jan C-5 Alarcon Damiana 89 26-Jun C-1 Albert Florence E. -

Provisional List of Participants
OSCE Human Dimension Implementation Meeting PROVISIONAL LIST OF PARTICIPANTS The Provisional List of Participants is based on registration sent to ODIHR Please submit any corrections/comments to the ODIHR Secretariat, Sofitel Victoria hotel or by e-mail to: [email protected] by Thursday, 3 October 2013, 2:00 p.m. Please note that any corrections provided later than Thursday, 3 October 2013, 2:00 p.m. will NOT be included in the Final List of Participants Warsaw, 23 September - 4 October 2013 OSCE Delegations / Partners for Co-operation Albania Amb. Spiro KOCI Permanent Mission of Albania to the International Organizations in Head of Delegation; Permanent Representative of the Republic of Albania to Vienna the OSCE Reisenerstrasse 27/6a; 1030 Vienna; Austria E-Mail: [email protected] Tel:+43-1-328 87 10 Website: http://www.mfa.gov.al Amb. Florent CELIKU Albanian Embassy to Poland E-Mail: [email protected] ul. Altowa 1; 02-386 Warsaw; Poland Tel:+48-22-824 14 27 Fax:+48-22-824 14 26 Ms. Selma XHOXHAJ Permanent Mission of Albania to the International Organizations in First Secretary Vienna E-Mail: [email protected] Reisenerstrasse 27/6a; 1030 Vienna; Austria Tel:+43-660-350 38 12 Website: http://www.mfa.gov.al Germany Amb. Rudiger LUDEKING Permanent Mission of the Federal Republic of Germany to the OSCE Head of Delegation Metternichgasse 3; 1030 Vienna; Austria E-Mail: [email protected] Tel:+43-1-711 54 171 Fax:+43-1-711 54 268 Website: http://www.osze.diplo.de Mrs. -

Wiezienie W Radomiu (Sygn. 417), 1939-1944
http://collections.ushmm.org Contact [email protected] for further information about this collection Wiezienie w Radomiu (Sygn. 417), 1939‐1944 The Prison in Radom‐Gefängins in Radom RG‐15.112M United States Holocaust Memorial Museum Archive 100 Raoul Wallenberg Place SW Washington, DC 20024‐2126 Tel. (202) 479‐9717 Email: [email protected] Descriptive Summary Title: Wiezienie w Radomiu (Sygn. 417) (The Prison in Radom‐Gefängins in Radom) Dates: 1939‐1944 RG Number: RG‐15.112M Accession Number: 2007.4 Extent: 22 microfilm reels (35 mm); 25,315 digital images (JPEG) Repository: United States Holocaust Memorial Museum Archive, 100 Raoul Wallenberg Place SW, Washington, DC 20024‐2126 Languages: German and Polish. Administrative Information Access: No restrictions on access. Reproduction and Use: Reproduction of more than 100 pages of copies of documents for researchers or other institutions requires a written permission of the General Director of the State Archives of the Republic of Poland. Publication of more than 10 complete documents in an individual work requires the written authorization of the General Director. The Museum may not publish any archival material obtained from the General Director, including specific archives under its control, on the Internet, the World Wide Web, or any other publicly accessible on‐line network without the written permission of the General Director. Citation of the materials in any publication must refer to the Museum and the Polish State Archives and must include the name of the archival group and catalogue number of the originals. 1 http://collections.ushmm.org http://collections.ushmm.org Contact [email protected] for further information about this collection To request written permission, contact the General Director, Naczelna Dyrekcja Archiwów Państwowych, ul. -

Fort Bend ISD from TEA Records As of July 8, 2008
Fort Bend ISD from TEA records as of July 8, 2008 first_name middle_name last_name test_descr admin_dt test_result test_score rolex Michelle Lee Aaronson-Starbuck Elementary Comprehensive 19-Nov-94 TEACHER Michelle Lee Aaronson-Starbuck Professional Development 19-Nov-94 DEPARTMENT HEAD Michelle Lee Aaronson-Starbuck Professional Development 19-Nov-94 TEACHER Ailynne Abarado English as a Second Language 04-Oct-03 TEACHER Ailynne Abarado Generalist EC-4 28-Jun-03 TEACHER Ailynne Abarado Pedagogy and Professional 06-Dec-03 TEACHER Amy Marie Abbott Professional Development (All- 12-Jun-93 TEACHER Amy Marie Abbott Special Education - Hearing 12-Jun-93 TEACHER Amy Marie Abbott Visually Handicapped 12-Jun-93 TEACHER Mary Aziz Abdelsayed Elementary Comprehensive 23-Feb-91 TEACHER Mary Aziz Abdelsayed English As A Second Language 26-Apr-97 TEACHER Barry Scott Abercrombie Composite Social Studies 25-Apr-98 SPECIAL DUTY TEACHER Barry Scott Abercrombie Composite Social Studies 25-Apr-98 TEACHER Barry Scott Abercrombie Health Education (Secondary) 17-Apr-99 TEACHER Barry Scott Abercrombie History (Secondary) 27-Apr-96 TEACHER Barry Scott Abercrombie Physical Education (Secondary) 27-Apr-96 TEACHER Barry Scott Abercrombie Professional Development 27-Apr-96 TEACHER Alvin V Abraham English as a Second Language 10-Dec-05 ASSISTANT PRINCIPAL Alvin V Abraham Generalist 4-8 08-Apr-06 ASSISTANT PRINCIPAL Alvin V Abraham Generalist EC-4 23-Nov-02 ASSISTANT PRINCIPAL Alvin V Abraham Pedagogy and Professional 01-Feb-03 ASSISTANT PRINCIPAL Alvin Varghese Abraham -

18 November 2016 to Cornell Interim
18 November 2016 To Cornell Interim President Rawlings and Incoming President Pollack: In this time of transition for Cornell University and our nation, and in keeping with Cornell’s mission of “any person any study,” we, the undersigned students, faculty, staff, and all members of the Cornell community, join students and faculty in colleges and universities across the nation in demanding that you honor and live up to these words by declaring Cornell University a sanctuary campus. Last week over 1,000 students organized, led, and participated in a walk out in response to the hate speech driving this year’s presidential election. Students stood up against every form of violence and oppression targeting marginalized people in this country and globally. They marched for “a world in which we can walk down the street without fearing for our lives and the lives of our loved ones.” Since last Friday, several of Cornell’s programs and departments have issued statements indicating their concern for the safety and well-being of the entire Cornell community. They have insisted that we maintain an environment in which all of our students can learn without fear. Meanwhile alumni, local leaders, and community members have also reaffirmed their support for human rights and dignity. Today we ask that you honor Ithaca’s tradition of being a place of sanctuary as well as Cornell’s historical roots in the abolition movement, by making an unequivocal statement of support for undocumented persons and their families. First, we ask you to explicitly denounce the acts of violence and hate speech directed at immigrant, ethnic and religious minorities, and LGBTQ students and workers. -
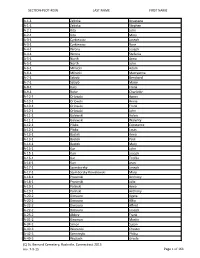
SECTION-PLOT-ROW LAST NAME FIRST NAME A-1-1 Zelinka
SECTION-PLOT-ROW LAST NAME FIRST NAME A-1-1 Zelinka Anastazia A-1-1 Zelinka Stephen A-2-1 Kita John A-2-1 Kita Mary A-3-1 Cyrkiewicz Joseph A-3-1 Cyrkiewicz Rose A-4-1 Wrona Joseph A-4-1 Wrona Stefania A-5-1 North Anna A-5-1 North John A-6-1 Milnicki Adam A-6-1 Milnicki Marcyanna A-7-1 Satryb Bernhard A-7-1 Satryb Marie A-8-1 Kulo Frank A-9-1 Rohe Charlotte A-10-1 Orlowski Agnes A-10-1 Orlowski Annie A-10-1 Orlowski Frank A-10-1 Orlowski John A-11-1 Galewski Helen A-11-1 Galewski Walenty A-12-1 Pliska Constance A-12-1 Pliska Louis A-13-1 Bastek Anna A-13-1 Bastek Paul A-14-1 Bastek Mary A-15-1 Kur John A-15-1 Kurr Joseph A-16-1 Kur Teofila A-16-1 Kurr Jean A-17-1 Szymborsky Joseph A-17-1 Szymborsky Kowalewski Mary A-18-1 Prosinski Anthony A-18-1 Prosinski Julia A-19-1 Polinski Anna A-19-1 Polinski Anthony A-20-2 Dimauro Agata A-20-2 Dimauro Alfio A-22-2 Dimauro Alfred A-22-2 Dimauro Joseph A-23-2 Abbey Frank A-31-2 Kincman Martin A-34-2 Simon Susan A-40-3 Wisnieski Chester A-42-3 Szemreylo Philca A-49-3 Wojtach Ursula (C) St. Bernard Cemetery, Rockville, Connecticut 2015 rev. 7-9-15 Page 1 of 161 SECTION-PLOT-ROW LAST NAME FIRST NAME A-53-3 Zastawny Stanislaw A-64-4 Unidentified A-65-4 Kalina Stanley A-68-4 Doran Ellen A-68-4 Doran Michael A-69-4 Griniewski Josephine A-69-4 Griniewski Teofil A-78-5 Sheehan James A-85-5 Orlowski Anna A-86-5 Kawalec Joseph A-87-5 Kulo Jacob A-87-5 Kulo Mary A-90-5 Kolman Nemeth A-93-5 Schenal Pietro A-97-6 Lokshig Joseph A-105-6 Kawalec Katherine A-107-6 Drenzek Catherine A-109-6 Dombek Catherine A-109-6 -

Noms De Famille Issus De L'artisanat En France Et En Pologne
ROCZNIKI HUMANISTYCZNE Tom LXVI, zeszyt 8 – 2019 DOI: http://dx.doi.org/10.18290/rh.2019.67.8-5 IWONA PIECHNIK1 NOMS DE FAMILLE ISSUS DE L’ARTISANAT EN FRANCE ET EN POLOGNE SURNAMES FROM ARTISAN NAMES IN FRANCE AND IN POLAND Abstract The article analyses surnames originating from artisan names in France and in Poland. It presents their origins (including foreign influences), types and word formation. We can see, among other things, that the French surnames are shorter, but have many dialectal variants, while the Polish surnames are longer and have a richer derivation. The article also focuses on demographic statis- tics of such surnames in both countries: the blacksmith as an etymon is the most popular. In the top 50, there are also in France: baker, miller and mason; while in Poland: tailor and shoemaker. Key words: family names; surnames; patronyms; handicraft; artisan. Les plus anciens noms de famille issus des domaines de l’artisanat en France et en Pologne remontent au Moyen Âge, donc à l’époque où le sys- tème féodal se renforçait et les villes commençaient à se développer, en nourrissant surtout les ambitions des nobles de construire leurs demeures seigneuriales, et des gens de petits métiers venaient s’installer tout autour naturellement. Dans des bourgs, c’est-à-dire dans de gros villages où se te- naient ordinairement des marchés, les bourgeois bénéficiaient d’un statut privilégié et développaient le commerce et la conjoncture de la manufacture, donc il y avait aussi beaucoup de travail pour différents métiers. C’est juste- ment dans les bourgs et les villes que l’artisanat se développait le mieux, en 1 Dr hab. -

Naturalization Index (1904-1958)
Naturalization Index (1904-1958) SURNAME FIRST NAMEMid Name VOL PAGE ADDRESS DATE ERTIFICATE MEMO Abbots William Lovatt 2 13 Buffington Twp 22-Feb-04 Auden Albert 2 47 Glen Campbell 19-Mar-04 Auden Alfred 2 81 Glen Campbell 19-Mar-04 Arur Pul 2 93 Glen Campbell 19-Mar-04 Alik Frank 2 109 Glen Campbell 28-Mar-04 Elik Anderson Andrew 2 149 Ernest 06-Oct-04 Areco Nicholas 2 175 Rossiter 18-Feb-06 Azzara Gaetano 1 7 Clymer 20-Aug-07 13972 Anderson Alfred 2A 16 Dixonville 16-Aug-10 46093 Adamson Charles 2A 33 Wehrum Denied (Later admitted) Adamson Charles 2A 45 Wehrum 27-Aug-11 192257 Adelburg Harry 2A 60 Iselin 13-Feb-12 236264 Alexander, John 2A 71 Dixonville 13-Feb-12 236268 Anderson Alex 3 12 Iselin 11-Feb-13 302444 Augustine Frank 3 98 Glen Campbell 19-Aug-13 406492 Adams John 4 5 Tunnelton 19-Aug-13 359826 Astalos John 4 35 Saltsburg 10-Feb-14 460511 Artzmowicz Frank 4 64 Clymer 18-Aug-14 460544 Andrasain Joseph 4 87 Saxman 09-Feb-15 516121 Anderson Abbonizio Consiglio 5 55 Tunnelton 04-Aug-15 501713 Abruzzi Massimo 5 100 Tunnelton Cancelled Alaimo Angelo 6 26 Chambersville 02-Feb-16 504783 Alimo Adair Hans, Jr. 6 67 Ernest 02-Aug-16 731877 Amantea Antonio 6 72 Heilwood 02-Aug-16 731872 Amerando Leonardo 7 2 McIntyre 15-Feb-17 735426 Abraham John 7 20 Clymer Spoiled petition Allesandro Vito 7 72 Chambersville 01-Aug-17 737682 Antonucci Donato 7 74 Strangford Denied Adamcik Imrich 8 24 Blairsville 05-May-20 1330954 Annunis Matt 8 43 Ernest 07-Feb-18 828258 Arvins Arvins Matt 8 43 Ernest 07-Feb-18 828258 Annunis Acciaccaferro Francesco