Kalea AZ Corrected Proof for Copyediting
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 7: Monogenic Forms of Diabetes
CHAPTER 7 MONOGENIC FORMS OF DIABETES Mark A. Sperling, MD, and Abhimanyu Garg, MD Dr. Mark A. Sperling is Emeritus Professor and Chair, University of Pittsburgh, Department of Pediatrics, Children’s Hospital of Pittsburgh of UPMC, Pittsburgh, PA. Dr. Abhimanyu Garg is Professor of Internal Medicine and Chief of the Division of Nutrition and Metabolic Diseases at University of Texas Southwestern Medical Center, Dallas, TX. SUMMARY Types 1 and 2 diabetes have multiple and complex genetic influences that interact with environmental triggers, such as viral infections or nutritional excesses, to result in their respective phenotypes: young, lean, and insulin-dependence for type 1 diabetes patients or older, overweight, and often manageable by lifestyle interventions and oral medications for type 2 diabetes patients. A small subset of patients, comprising ~2%–3% of all those diagnosed with diabetes, may have characteristics of either type 1 or type 2 diabetes but have single gene defects that interfere with insulin production, secretion, or action, resulting in clinical diabetes. These types of diabetes are known as MODY, originally defined as maturity-onset diabetes of youth, and severe early-onset forms, such as neonatal diabetes mellitus (NDM). Defects in genes involved in adipocyte development, differentiation, and death pathways cause lipodystrophy syndromes, which are also associated with insulin resistance and diabetes. Although these syndromes are considered rare, more awareness of these disorders and increased availability of genetic testing in clinical and research laboratories, as well as growing use of next generation, whole genome, or exome sequencing for clinically challenging phenotypes, are resulting in increased recognition. A correct diagnosis of MODY, NDM, or lipodystrophy syndromes has profound implications for treatment, genetic counseling, and prognosis. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Evidence Revealing Deregulation of the KLF11-Mao a Pathway in Association with Chronic Stress and Depressive Disorders
Neuropsychopharmacology (2015) 40, 1373–1382 & 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 www.neuropsychopharmacology.org Evidence Revealing Deregulation of The KLF11-Mao A Pathway in Association with Chronic Stress and Depressive Disorders 1 1 1,2 1 3 Sharonda Harris , Shakevia Johnson , Jeremy W Duncan , Chinelo Udemgba , Jeffrey H Meyer , Paul R Albert4, Gwen Lomberk5, Raul Urrutia5, Xiao-Ming Ou1, Craig A Stockmeier1,6 and Jun Ming Wang*,1,2,7 1 2 3 Department of Psychiatry and Human Behavior, Jackson, MS, USA; Program in Neuroscience, Jackson, MS, USA; Centre for Addiction and Mental Health and Department of Psychiatry, University of Toronto, Toronto, Ontario, Canada; 4Ottawa Hospital Research Institute (Neuroscience), Ottawa, Ontario, Canada; 5Epigenetics and Chromatin Dynamics Laboratory, GI Research Unit, Mayo Clinic, Rochester, MN, 6 7 USA; Department of Psychiatry, Case Western Reserve University, Cleveland, OH, USA; Department of Pathology, University of Mississippi Medical Center, Jackson, MS, USA The biochemical pathways underlying major depressive disorder (MDD) and chronic stress are not well understood. However, it has been reported that monoamine oxidase A (MAO A, a major neurotransmitter-degrading enzyme) is significantly increased in the brains of human subjects affected with MDD and rats exposed to chronic social defeat (CSD) stress, which is used to model depression. In the current study, we compared the protein levels of a MAO A-transcriptional activator, Kruppel-like factor 11 (KLF11 , also recognized as transforming growth factor-beta-inducible early gene 2) between the brains of 18 human subjects with MDD and 18 control subjects. We found that, indeed, the expression of KLF11 is increased by 36% (po0.02) in the postmortem prefrontal cortex of human subjects with MDD compared with controls. -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals
animals Review Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals Mohammadreza Mohammadabadi 1 , Farhad Bordbar 1,* , Just Jensen 2 , Min Du 3 and Wei Guo 4 1 Department of Animal Science, Faculty of Agriculture, Shahid Bahonar University of Kerman, Kerman 77951, Iran; [email protected] 2 Center for Quantitative Genetics and Genomics, Aarhus University, 8210 Aarhus, Denmark; [email protected] 3 Washington Center for Muscle Biology, Department of Animal Sciences, Washington State University, Pullman, WA 99163, USA; [email protected] 4 Muscle Biology and Animal Biologics, Animal and Dairy Science, University of Wisconsin-Madison, Madison, WI 53558, USA; [email protected] * Correspondence: [email protected] Simple Summary: Skeletal muscle mass is an important economic trait, and muscle development and growth is a crucial factor to supply enough meat for human consumption. Thus, understanding (candidate) genes regulating skeletal muscle development is crucial for understanding molecular genetic regulation of muscle growth and can be benefit the meat industry toward the goal of in- creasing meat yields. During the past years, significant progress has been made for understanding these mechanisms, and thus, we decided to write a comprehensive review covering regulators and (candidate) genes crucial for muscle development and growth in farm animals. Detection of these genes and factors increases our understanding of muscle growth and development and is a great help for breeders to satisfy demands for meat production on a global scale. Citation: Mohammadabadi, M.; Abstract: Farm-animal species play crucial roles in satisfying demands for meat on a global scale, Bordbar, F.; Jensen, J.; Du, M.; Guo, W. -
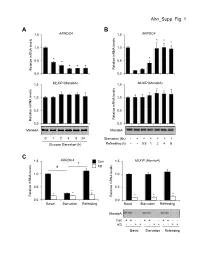
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

Open Chromatin Profiling in Adipose Tissue Marks Genomic Regions with Functional Roles in Cardiometabolic Traits
INVESTIGATION Open Chromatin Profiling in Adipose Tissue Marks Genomic Regions with Functional Roles in Cardiometabolic Traits Maren E. Cannon,*,1 Kevin W. Currin,*,1 Kristin L. Young,† Hannah J. Perrin,* Swarooparani Vadlamudi,* Alexias Safi,‡ Lingyun Song,‡ Ying Wu,* Martin Wabitsch,§ Markku Laakso,** Gregory E. Crawford,‡ and Karen L. Mohlke*,2 *Department of Genetics, †Department of Epidemiology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, ‡Department of Pediatrics, Division of Medical Genetics and Center for Genomic and Computational Biology, § Duke University, NC 27710, Department of Pediatrics and Adolescents Medicine, Division of Pediatric Endocrinology and Diabetes, University of Ulm, Germany 89081, and **Department of Medicine, University of Eastern Finland and Kuopio University Hospital, Kuopio, Finland 70210 ORCID ID: 0000-0001-6721-153X (K.L.M.) ABSTRACT Identifying the regulatory mechanisms of genome-wide association study (GWAS) loci KEYWORDS affecting adipose tissue has been restricted due to limited characterization of adipose transcriptional chromatin regulatory elements. We profiled chromatin accessibility in three frozen human subcutaneous adipose accessibility tissue needle biopsies and preadipocytes and adipocytes from the Simpson Golabi-Behmel Syndrome adipose tissue (SGBS) cell strain using an assay for transposase-accessible chromatin (ATAC-seq). We identified preadipocytes 68,571 representative accessible chromatin regions (peaks) across adipose tissue samples (FDR , 5%). GWAS GWAS loci for eight cardiometabolic traits were enriched in these peaks (P , 0.005), with the strongest cardiometabolic enrichment for waist-hip ratio. Of 110 recently described cardiometabolic GWAS loci colocalized with trait adiposetissueeQTLs,59locihadoneormorevariantsoverlappinganadiposetissuepeak.Annotated variants at the SNX10 waist-hip ratio locus and the ATP2A1-SH2B1 body mass index locus showed allelic differences in regulatory assays. -

Tgfβ-Regulated Gene Expression by Smads and Sp1/KLF-Like Transcription Factors in Cancer VOLKER ELLENRIEDER
ANTICANCER RESEARCH 28 : 1531-1540 (2008) Review TGFβ-regulated Gene Expression by Smads and Sp1/KLF-like Transcription Factors in Cancer VOLKER ELLENRIEDER Signal Transduction Laboratory, Internal Medicine, Department of Gastroenterology and Endocrinology, University of Marburg, Marburg, Germany Abstract. Transforming growth factor beta (TGF β) controls complex induces the canonical Smad signaling molecules which vital cellular functions through its ability to regulate gene then translocate into the nucleus to regulate transcription (2). The expression. TGFβ binding to its transmembrane receptor cellular response to TGF β can be extremely variable depending kinases initiates distinct intracellular signalling cascades on the cell type and the activation status of a cell at a given time. including the Smad signalling and transcription factors and also For instance, TGF β induces growth arrest and apoptosis in Smad-independent pathways. In normal epithelial cells, TGF β healthy epithelial cells, whereas it can also promote tumor stimulation induces a cytostatic program which includes the progression through stimulation of cell proliferation and the transcriptional repression of the c-Myc oncogene and the later induction of an epithelial-to-mesenchymal transition of tumor induction of the cell cycle inhibitors p15 INK4b and p21 Cip1 . cells (1, 3). In the last decade it has become clear that both the During carcinogenesis, however, many tumor cells lose their tumor suppressing and the tumor promoting functions of TGF β ability to respond to TGF β with growth inhibition, and instead, are primarily regulated on the level of gene expression through activate genes involved in cell proliferation, invasion and Smad-dependent and -independent mechanisms (1, 2, 4). -

KLF11 Protects Against Abdominal Aortic Aneurysm Through Inhibition of Endothelial Cell Dysfunction
KLF11 protects against abdominal aortic aneurysm through inhibition of endothelial cell dysfunction Guizhen Zhao, … , Eugene Chen, Jifeng Zhang JCI Insight. 2021. https://doi.org/10.1172/jci.insight.141673. Research In-Press Preview Vascular biology Graphical abstract Find the latest version: https://jci.me/141673/pdf 1 KLF11 protects against abdominal aortic aneurysm through inhibition of endothelial cell 2 dysfunction 3 Guizhen Zhao1, Ziyi Chang1,2, Yang Zhao1, Yanhong Guo1, Haocheng Lu1, Wenying Liang1, 4 Oren Rom1, Huilun Wang1, Jinjian Sun1,3, Tianqing Zhu1, Yanbo Fan1,4, Lin Chang1, Bo Yang5, 5 Minerva T. Garcia-Barrio1, Y. Eugene Chen1,5 and Jifeng Zhang1 6 7 Affiliations: 1Department of Internal Medicine, University of Michigan Medical Center, Ann 8 Arbor, MI 48109; 2Department of Pathology, China-Japan Friendship Hospital, Beijing 100029, 9 China ; 3Department of Cardiovascular Medicine, the Second Xiangya Hospital, Central South 10 University, Changsha, Hunan 410011, China; 4Department of Cancer Biology, College of 11 Medicine, University of Cincinnati, Cincinnati, OH 45267; 5Department of Cardiac Surgery, 12 University of Michigan Medical Center, Ann Arbor, MI 48109. 13 Corresponding Authors: 14 Jifeng Zhang, PhD, Department of Internal Medicine, University of Michigan Medical Center, 15 NCRC Bldg26-357S. 2800 Plymouth Rd, Ann Arbor, MI 48109, Email: [email protected] 16 Y. Eugene Chen, MD, PhD, Department of Internal Medicine, University of Michigan Medical 17 Center, NCRC Bldg26-361S. 2800 Plymouth Rd, Ann Arbor, MI 48109, Email: 18 [email protected] 19 The authors have declared that no conflict of interest exists. 20 1 1 Abstract 2 Abdominal aortic aneurysm (AAA) is a life-threatening degenerative vascular disease. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Immune Suppressive Landscape in the Human Esophageal Squamous Cell
ARTICLE https://doi.org/10.1038/s41467-020-20019-0 OPEN Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment ✉ Yingxia Zheng 1,2,10 , Zheyi Chen1,10, Yichao Han 3,10, Li Han1,10, Xin Zou4, Bingqian Zhou1, Rui Hu5, Jie Hao4, Shihao Bai4, Haibo Xiao5, Wei Vivian Li6, Alex Bueker7, Yanhui Ma1, Guohua Xie1, Junyao Yang1, ✉ ✉ ✉ Shiyu Chen1, Hecheng Li 3 , Jian Cao 7,8 & Lisong Shen 1,9 1234567890():,; Cancer immunotherapy has revolutionized cancer treatment, and it relies heavily on the comprehensive understanding of the immune landscape of the tumor microenvironment (TME). Here, we obtain a detailed immune cell atlas of esophageal squamous cell carcinoma (ESCC) at single-cell resolution. Exhausted T and NK cells, regulatory T cells (Tregs), alternatively activated macrophages and tolerogenic dendritic cells are dominant in the TME. Transcriptional profiling coupled with T cell receptor (TCR) sequencing reveal lineage con- nections in T cell populations. CD8 T cells show continuous progression from pre-exhausted to exhausted T cells. While exhausted CD4, CD8 T and NK cells are major proliferative cell components in the TME, the crosstalk between macrophages and Tregs contributes to potential immunosuppression in the TME. Our results indicate several immunosuppressive mechanisms that may be simultaneously responsible for the failure of immuno-surveillance. Specific targeting of these immunosuppressive pathways may reactivate anti-tumor immune responses in ESCC. 1 Department of Laboratory Medicine, Xin Hua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 2 Institute of Biliary Tract Diseases Research, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 3 Department of Thoracic Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China. -

Metabolic Profiling of Cancer Cells Reveals Genome-Wide
ARTICLE https://doi.org/10.1038/s41467-019-09695-9 OPEN Metabolic profiling of cancer cells reveals genome- wide crosstalk between transcriptional regulators and metabolism Karin Ortmayr 1, Sébastien Dubuis1 & Mattia Zampieri 1 Transcriptional reprogramming of cellular metabolism is a hallmark of cancer. However, systematic approaches to study the role of transcriptional regulators (TRs) in mediating 1234567890():,; cancer metabolic rewiring are missing. Here, we chart a genome-scale map of TR-metabolite associations in human cells using a combined computational-experimental framework for large-scale metabolic profiling of adherent cell lines. By integrating intracellular metabolic profiles of 54 cancer cell lines with transcriptomic and proteomic data, we unraveled a large space of associations between TRs and metabolic pathways. We found a global regulatory signature coordinating glucose- and one-carbon metabolism, suggesting that regulation of carbon metabolism in cancer may be more diverse and flexible than previously appreciated. Here, we demonstrate how this TR-metabolite map can serve as a resource to predict TRs potentially responsible for metabolic transformation in patient-derived tumor samples, opening new opportunities in understanding disease etiology, selecting therapeutic treat- ments and in designing modulators of cancer-related TRs. 1 Institute of Molecular Systems Biology, ETH Zurich, Otto-Stern-Weg 3, CH-8093 Zurich, Switzerland. Correspondence and requests for materials should be addressed to M.Z. (email: [email protected]) NATURE COMMUNICATIONS | (2019) 10:1841 | https://doi.org/10.1038/s41467-019-09695-9 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-019-09695-9 ranscriptional regulators (TRs) are at the interface between implemented by the additional quantification of total protein the cell’s ability to sense and respond to external stimuli or abundance and is based on the assumption that protein content T 1 changes in internal cell-state .