The Cells of the Oligodendrocyte Lineage Are Differentially Altered by Tau Accumulation and Amyloidosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
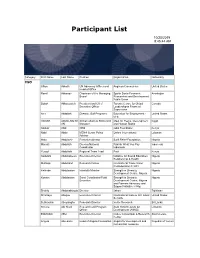
Participant List
Participant List 10/20/2019 8:45:44 AM Category First Name Last Name Position Organization Nationality CSO Jillian Abballe UN Advocacy Officer and Anglican Communion United States Head of Office Ramil Abbasov Chariman of the Managing Spektr Socio-Economic Azerbaijan Board Researches and Development Public Union Babak Abbaszadeh President and Chief Toronto Centre for Global Canada Executive Officer Leadership in Financial Supervision Amr Abdallah Director, Gulf Programs Educaiton for Employment - United States EFE HAGAR ABDELRAHM African affairs & SDGs Unit Maat for Peace, Development Egypt AN Manager and Human Rights Abukar Abdi CEO Juba Foundation Kenya Nabil Abdo MENA Senior Policy Oxfam International Lebanon Advisor Mala Abdulaziz Executive director Swift Relief Foundation Nigeria Maryati Abdullah Director/National Publish What You Pay Indonesia Coordinator Indonesia Yussuf Abdullahi Regional Team Lead Pact Kenya Abdulahi Abdulraheem Executive Director Initiative for Sound Education Nigeria Relationship & Health Muttaqa Abdulra'uf Research Fellow International Trade Union Nigeria Confederation (ITUC) Kehinde Abdulsalam Interfaith Minister Strength in Diversity Nigeria Development Centre, Nigeria Kassim Abdulsalam Zonal Coordinator/Field Strength in Diversity Nigeria Executive Development Centre, Nigeria and Farmers Advocacy and Support Initiative in Nig Shahlo Abdunabizoda Director Jahon Tajikistan Shontaye Abegaz Executive Director International Insitute for Human United States Security Subhashini Abeysinghe Research Director Verite -

Petition Signatories 12 November 2020 First Name Surname Country Capacity
Petition Signatories 12 November 2020 First name Surname Country Capacity 1 Ulisses Abade Brasil Vice Presidente Sindicato 2 Sandrine Abayou France salariée 3 HAYANI ABDEL BELGIUM trade union 4 Ariadna Abeltina Latvia Trade Union Officer General Secretary FSC 5 Roberto Abenia España CCOO Aragón 6 Pascal Abenza France Délégué Syndical Groupe 7 Jacques ADAM Luxembourg membre du syndicat 8 Lenka Adamcikova Slowakei Member of works 9 Ole Einar Adamsrød Norway Trade Union 10 Paula Adao Luxembourg Déléguée 11 Nicolae Adrian România Union member 12 Costache Adrian Alin Romania Trade union 13 Pana Adriana Laura România Member of works council 14 Bert Aerts Belgium member of works council 15 Annick Aerts Belgium trade union 16 Sascha Aerts België 200 Secrétaire Générale UL 17 Odile AGRAFEIL FRANCE CGT 18 Oscar Aguado España Miembro comité empresa 19 Fátima Aguado Queipo Spain Trade union 20 Agustin Aguila Mellado España Miembro del Sindicato SECRETARIO UGT ADIF 21 HERNANDEZ AGUILAR OSWALD BARCELONA 22 Antonio Angel Aguilar Fernández España Trade Union 23 Jaan Aiaots Estonia Trade union SYNDICAT SYNPTAC-CGT 24 Nora AINECHE FRANCE PARIS FRANCE 25 Raul Aira España miembro comité empresa 26 Alessandra Airaldi Italy TRADE union 27 Juan Miguel Aisa Spain Member of works council 28 Juan-Miguel AISA Spain EWC Membre élu du comité européen Driver Services 29 Sylvestre AISSI France Norauto 30 Sara Akervall Sweden EWC 31 Michiel Al Netherlands trade union official 32 Nickels Alain Luxemburg Trade Union 33 MAURO ALBANESE FRANCE SYNDICAT 34 Michela Albarello -

Diplomatic List – Fall 2018
United States Department of State Diplomatic List Fall 2018 Preface This publication contains the names of the members of the diplomatic staffs of all bilateral missions and delegations (herein after “missions”) and their spouses. Members of the diplomatic staff are the members of the staff of the mission having diplomatic rank. These persons, with the exception of those identified by asterisks, enjoy full immunity under provisions of the Vienna Convention on Diplomatic Relations. Pertinent provisions of the Convention include the following: Article 29 The person of a diplomatic agent shall be inviolable. He shall not be liable to any form of arrest or detention. The receiving State shall treat him with due respect and shall take all appropriate steps to prevent any attack on his person, freedom, or dignity. Article 31 A diplomatic agent shall enjoy immunity from the criminal jurisdiction of the receiving State. He shall also enjoy immunity from its civil and administrative jurisdiction, except in the case of: (a) a real action relating to private immovable property situated in the territory of the receiving State, unless he holds it on behalf of the sending State for the purposes of the mission; (b) an action relating to succession in which the diplomatic agent is involved as an executor, administrator, heir or legatee as a private person and not on behalf of the sending State; (c) an action relating to any professional or commercial activity exercised by the diplomatic agent in the receiving State outside of his official functions. -- A diplomatic agent’s family members are entitled to the same immunities unless they are United States Nationals. -

2019 World University Games: Men's Psych Sheet
Piscina Scandone Swimming Men's 50m Freestyle Entry List N. SURNAME & NAME CTRY BORN UNIVERSITY Q.TIME Event 33 09 JUL 2019 1 SENDYK Pawel POL 24 MAY 1997 U. Of California Berkeley 21.91 2 CUMBERLIDGE David Ross GBR 16 JUL 1996 Heriot-Watt University 22.00 3 JENSEN Michael USA 11 DEC 1997 U. Of California Berkeley 22.03 4 MATSUI Kosuke JPN 28 MAR 1994 Niigata U. Of Health & Welfare 22.06 5 APPLE Zachary USA 23 APR 1997 Indiana University Bloomington 22.06 6 WADDELL Zane RSA 18 MAR 1998 University Of Alabama 22.07 7 KUZMENKO Ivan RUS 07 APR 1995 Saratov State Agrarian Vavilov 22.12 8 SAKAI Koshiro JPN 27 OCT 1996 Chuo University 22.13 9 BORGES Luiz Gustavo BRA 21 MAY 1999 University Of Michigan 22.23 10 HO Ian Yentou HKG 25 APR 1997 University Virginia 22.25 11 STOCKWELL William AUS 30 JUL 1995 The University Of Queensland 22.29 12 BELL Grayson AUS 21 MAR 1997 Griffith University 22.30 13 VARAKIN Alexandr KAZ 17 FEB 1996 Kazakh A. Of Sport And Tourism 22.31 14 KSIAZEK Jakub POL 02 APR 1998 Florida State University 22.31 15 LATKIN Anton BLR 21 FEB 1994 Polotsk State University 22.44 16 AYOUBI Mehdi CAN 06 AUG 1998 Cegep A Distance 22.50 17 TEONG Tzen Wei SGP 17 OCT 1997 Singapore Management University 22.52 18 DE SOUZA Felipe BRA 19 FEB 1998 Anhanguera Centre Of Sao Paulo 22.53 19 MARKOV Daniil RUS 21 APR 2000 Novosibirsk State Pedagogical 22.53 20 YANG Jaehoon KOR 07 MAY 1998 Jeju International University 22.55 21 GIGLER Heiko AUT 17 JUN 1996 University Of Graz 22.56 22 BORI Alessandro ITA 05 FEB 1997 Pegaso Telematic University 22.59 23 ZOULALIAN Nicolas SUI 30 MAR 1996 University Of Lausanne 22.60 24 OSTROWSKI Karol POL 02 SEP 1999 Szczecin University 22.65 25 HUNTER Daniel NZL 28 JUN 1994 Massey U. -

Catholic Directory
CATHOLIC DIRECTORY ' M OF INDIA, rAXLSTArt, B uRm a * tfb C £ Yl a- tf< 1922 72nd ANNUAL ISSUE OF THE MADRAS CATHOLIC DIRECTORY AND ANNUAL GENERAL REGISTER PUBLISHED BY THE CATHOLIC SUPPLY SOCIETY, MADRAS. PRINTED AT THE “ GOOD PASTOR ” PRESS, BROADWAY, MADRAS, M T +Z / , 7 1 Nihil obstet : J. BEUKERS, Censor Deputatus. Imprimatur : * J. AELEN, Archiepiscopus Madraspatanus. Madras, die 21a mensis Decembris, 1921. PREFACE Another year has been added to the cen turies buried in the past, another year has been ushered in by joyful hymns. It is the old, old story. On the threshold of the new year we always resolve to spend the ensuing one better in the light we gained during the preceding twelve months. It is the old, old story. The 1921 edition of the Catholic Directory of India, Burma and Ceylon was far from complete, and the respective Chancellarles seeing this resolved no doubt that 1922 would see a copy unheard of for accuracy even in the life of this useful publication. All the ’ reports came in. Not one point—if we except Statistics—for the Compiler to complain about. To all and every one our best thanks. Y et there is in the present issue one omis sion which we regret. Three times we wrote asking for a photo and a brief sketch of the ^ new Vicar Apostolic of Trichur, and we were disappointed not to receive either in time for insertion. The notes inserted will be appreciated by all our readers. The contributors are heartily thanked for these sketches. It may be recorded that the Compiler is grateful for any useful suggestion. -

Free Tips for Searching Ancestors' Surnames
SURNAMES: FAMILY SEARCH TIPS AND SURNAME ORIGINS Picking a name Naming practices developed differently from region to region and country to country. Yet even today, hereditary The Name Game names tend to fall into one of four categories: patronymic Onomastics, a field of linguistics, is the study of names and (named from the father), occupational, nickname or place naming practices. The American Name Society (ANS) was name. According to Elsdon Smith, author of American Sur- founded in 1951 to promote this field in the United States and names (Genealogical Publishing Co.), a survey of some 7,000 abroad. Its goal is to “find out what really is in a name, and to surnames in America revealed that slightly more than 43 investigate cultural insights, settlement history and linguistic percent of our names derive from places, followed by about characteristics revealed in names.” 32 percent from patronymics, 15 percent from occupations The society publishes NAMES: A Journal of Onomastics, and 9 percent from nicknames. a quarterly journal; the ANS Bulletin; and the Ehrensperger Often the lines blur between the categories. Take the Report, an annual overview of member activities in example of Green. This name could come from one’s clothing onomastics. The society also offers an online discussion or it could be given to one who was inexperienced. It could group, ANS-L. For more information, visit the ANS website at also mean a dweller near the village green, be a shortened <www.wtsn.binghamton.edu/ANS>. form of a longer Jewish or German name, or be a translation from another language. -

ELI Fellows by Surname
ELI Fellows by Surname Surname: First Name(s): Country: Abatangelo Chiara Italy Abbiati Paul Portugal Åbjörnsson Rolf Sweden Abreu Joana Portugal Abu Awwad Amal Italy Achache Florence France Adame Martínez Miguel Ángel Spain Addante Adriana Italy Adler Peter H. Austria Afanasyeva Ekaterina Russia Afferni Giorgio Italy Agudo Gonzalez Jorge Spain Aguilera Marien Spain Ahrens Hans-Jürgen Germany Aichberger Beig Daphne Austria Aimo Mariapaola Italy Akkermans Bram Netherlands Akseli N. Orkun United Kingdom Alba Fernandez Manuel Spain Albert Maria Rosario Spain Alberti Lucia Giuseppina Italy Alemanno Alberto France Alexandropoulou Antigoni Cyprus Alexandru Aurelian Chirita Romania Alexe Alina Romania Allemeersch Benoît Belgium Aloj Nicoletta Italy Alonso Landeta Gabriel Spain Alonso Perez Mª Teresa Spain Alpa Guido Italy Alunaru Christian Romania Amin-Mannion Rosy United Kingdom Amodio Claudia Italy Amtenbrink Fabian Netherlands Anagnostopoulos Ilias Greece Anagnostopoulou Despoina Greece Anches Diana-Ionela Romania Andenas Mads Norway Anderson Ross United Kingdom Andrews Neil United Kingdom Androulakis Ioannis Greece Anker-Sørensen Linn Norway Annoni Alessandra Italy Anthimos Apostolos Greece Antoniolli Luisa Italy Appert Geraldine United Kingdom Arabadjiev Alexander Luxembourg Aran Latif Cyprus Arastey Maria Lourdes Spain Armeli Beatrice Italy Armenta Deu Teresa Spain Armstrong Kenneth United Kingdom Arnold Rainer Germany Arroyo Tatiana Spain Arroyo Amayuelas Esther Spain Atamer Yesim M. Turkish Republic Aubert de Vincelles Carole France -

BEHAVIORAL HEALTH PROVIDERS (Effective 1/1/2012) for More Information About Any Providers Listed Below, Please Call Our Member Services Department at 877-492-6967
BMC HEALTHNET PLAN SELECT -- BEHAVIORAL HEALTH PROVIDERS (effective 1/1/2012) For more information about any providers listed below, please call our Member Services department at 877-492-6967. Office Street Facility Name Last Name First Name Address Office Suite City State Zip Office Phone Medical Group Affiliation AdCare Hospital of Worcester, AdCare Hospital of Worcester, Inc. - Worcester Site 107 Lincoln Street Worcester MA 01605 (800)345-3552 Inc. - Worcester Site Arbour Hospital 49 Robinwood Ave Boston MA 02130 (617)522-4400 Arbour Hospital Dimock Detox 55 Dimock Street Roxbury MA 02119 (617)442-9661 Dimock Detox Arbour HRI Hospital 227 Babcock St. Brookline MA 02146 (617)731-3200 Arbour HRI Hospital Child and Family Services, Inc. 1061 Pleasant Street New Bedford MA 02740 (508)996-8572 Child and Family Services, Inc. Child and Family Services, Inc. 1061 Pleasant Street New Bedford MA 02740 (508)996-8572 Child and Family Services, Inc. Bayridge Hospital 60 Granite Street Lynn MA 01904 (781)599-9200 Bayridge Hospital Bournewood Hospital 300 South Street Brookline MA 02467 (617)469-0300 Bournewood Hospital Bournewood Hospital 300 South Street Brookline MA 02467 (617)469-0300 Bournewood Hospital 1493 Cambridge Cambridge Hospital Street Cambridge MA 02139 (617)498-1000 Cambridge Hospital Community Healthlink - 72 Jaques Community Healthlink - 72 Avenue 72 Jaques Avenue Worcester MA 01610 (508)860-1260 Jaques Avenue Faulkner Hospital 1153 Center Street Jamaica Plain MA 02130 (617)983-7711 Faulkner Hospital Lowell Community Health Lowell Community Health Initiative 15-17 Warren Street Lowell MA 01852 (978)937-9448 Initiative McLean Hospital 115 Mill St. -
Participant List
Participant List 10/20/2020 12:59:08 PM Category First Name Last Name Position Organization Nationality CSO Jamal Aazizi Chargé de la logistique Association Tazghart Morocco Luz Abayan Program Officer Child Rights Coalition Asia Philippines Babak Abbaszadeh President And Chief Toronto Centre For Global Canada Executive Officer Leadership In Financial Supervision Amr Abdallah Director, Gulf Programs Education for Employment - United States EFE Ziad Abdel Samad Executive Director Arab NGO Network for Lebanon Development TAZI Abdelilah Président Associaion Talassemtane pour Morocco l'environnement et le développement ATED Abla Abdellatif Executive Director and The Egyptian Center for Egypt Director of Research Economic Studies Nabil Abdo MENA Senior Policy Oxfam International Lebanon Advisor Baako Abdul-Fatawu Executive Director Centre for Capacity Ghana Improvement for the Wellbeing of the Vulnerable (CIWED) Maryati Abdullah Director/National Publish What You Pay Indonesia Coordinator Indonesia Dr. Abel Executive Director Reach The Youth Uganda Switzerland Mwebembezi (RTY) Suchith Abeyewickre Ethics Education Arigatou International Sri Lanka me Programme Coordinator Diam Abou Diab Fellow Arab NGO Network for Lebanon Development Hayk Abrahamyan Community Organizer for International Accountability Armenia South Caucasus and Project Central Asia Aliyu Abubakar Secretary General Kano State Peace and Conflict Nigeria Resolution Association Sunil Acharya Regional Advisor, Climate Practical Action Nepal and Resilience Salim Adam Public Health -

Start List 30 June 2021 21 November 2021 Age Group
START LIST 30 JUNE 2021 21 NOVEMBER 2021 AGE GROUP NAME SURNAME FEMALES F18-24 Elnae Neethling F25-29 Melissa Abbey F25-29 Jessica De Lange F25-29 Christine Harding F25-29 Nicola Janse Van Rensburg F25-29 Marjolein Mansvelder F25-29 Gena Nys F25-29 Taneal Otto F25-29 Kyla Purdon F25-29 Meagan Rioux F25-29 Sanri Steyn F25-29 Michaela Van Den Honert F25-29 Elicia Venter F30-34 Nina Ain El Fitre F30-34 Aisha Ameen F30-34 Lauren Bartle F30-34 Beulah Bekker F30-34 Chanelle Birch F30-34 Adelle Brits F30-34 Andrea Buhr F30-34 Michelle Cremer F30-34 Ione De Vos F30-34 Risa Dreyer F30-34 Shea Duncan F30-34 Francesca Ford F30-34 Polina Gryaznova F30-34 Jessica Hamuy Blanco F30-34 Lindsay Jolly F30-34 Elzani Lotter F30-34 Maxine Luck F30-34 Elise Meyering F30-34 Carolina Militão F30-34 Rosalind Netto F30-34 Megan Newman F30-34 Antoinette Niehaus F30-34 Elmi Nieuwoudt F30-34 Ayesha Osman F30-34 Kirsten Schut F30-34 Jo Shadwell F30-34 Paula Somerville F30-34 Karin Stapelberg F30-34 Caitlin Tracey F30-34 Kaylin Verreyne F35-39 Natalie Agostinho F35-39 Johesta Aspeling F35-39 Kerstin Bannwolf F35-39 Melanie Beets F35-39 Natasha Boshoff F35-39 Jacqui Bunge F35-39 Tracy Dennis F35-39 Terene Du Plessis F35-39 Pauline Emauere F35-39 Kaleo Fourie F35-39 Rebecca Gatang'I F35-39 Rebecca Goodwin F35-39 Eulali Gouws F35-39 Katherine Hardy F35-39 Samantha Harper F35-39 Lindi Humphreys F35-39 Sannelize Janse Van Rensburg F35-39 Anita Kaumpek F35-39 Tatyana Kosareva F35-39 Berber Kramer F35-39 Linelda Kühn F35-39 Manri Lourens F35-39 Shannon Lourens F35-39 Rianna -

CATHOLIC DIRECTORY « (T OF
CATHOLIC DIRECTORY « (t OF INDIA, FArtJ-STAXj I5u#/va A y y p c EY l o N' 1925 75th ANNUAL ISSUE OF THE MADRAS CATHOLIC DIRECTORY AND ANNUAL GENERAL REGISTER PUBLISHED BY THE CATHOLIC SUPPLY SOCIETY, MADRAS. PRINTED AT THE “ GOOD PASTOR ” PRESS, BROADWAY, MADRAS. file JHvinity Library to# Haven, Conn. M T ^ f ? C « 2 S i T iA 7 6 - , Nihil obstat. A. F. THEODORE, Censor Deputatus. Imprimatur: * J. AELEN, Archbishop o f Madras Madras, 17th December 1924. PREFACE. This, the Seventy-Fifth issue of the Catholic Directory of India, Burma and Ceylon, goes out under the happy auspices of a Blessing from the Visitor Apostolic to India. His Excellency the Most Revel. Alexis H. Lepicier, O.S.M., D D ., has very graciously blessed the Compiler and all those who have co-operated with him in bringing out the Directory. That this issue has merited in any special manner this courteous treat ment, is, to the Compiler, a matter of doubt, for he is fully aware that in spite of every effort at accuracy, errors and misprints have crept in. Yet, he feels sure that this Volume, like its, renowned predecessors, goes forth on its annual mission of usefulness and interest to a large number of Catholics of India, Burma and Ceylon. In the name of his Co-operators and in his own name, the Compiler thanks His Excellency the Visitor Apostolic for his very kind thought and Blessing. s It is a pleasant duty also to the Compiler to thank all those who have helped him in getting out this issue, though he has also to witzs/on confess that a few have failed in submitting >the usual sacred returns. -

PSE: Castex, Ferreira Elisa UEN: Aylward, Crowley, Ó Neachtain
C 157 E/214 Journal officiel de l'Union européenne FR 6.7.2006 Mercredi, 6 juillet 2005 PSE: Castex, Ferreira Elisa UEN: Aylward, Crowley, Ó Neachtain, Pavilionis, Ryan, Vaidere Verts/ALE: van Buitenen 48. Rapport Casa A6-0217/2005 Proposition Commission Pour: 481 ALDE: Andria, Beaupuy, Bonino, Bourlanges, Cavada, Cocilovo, Cornillet, Costa, Degutis, Deprez, Ek, Fourtou, Geremek, Gibault, Griesbeck, Guardans Cambó, Kułakowski, Laperrouze, Lehideux, Letta, Mohácsi, Morillon, Neyts-Uyttebroeck, Onyszkiewicz, Ortuondo Larrea, Pistelli, Prodi, Ries, Sbarbati, Schuth, Szent-Iványi, Toia, Virrankoski GUE/NGL: Liotard, Meijer, Seppänen, Sjöstedt, Svensson IND/DEM: Belder, Blokland, Chruszcz, Coûteaux, Giertych, Grabowski, Karatzaferis, Krupa, Louis, Pęk, Piotrowski, Rogalski, Salvini, Sinnott, Tomczak, de Villiers NI: Battilocchio, Belohorská, Claeys, Czarnecki Marek Aleksander, Czarnecki Ryszard, Dillen, Gollnisch, Lang, Le Pen Jean-Marie, Le Pen Marine, Le Rachinel, Martinez, Masiel, Mussolini, Rivera, Romagnoli, Rutowicz, Vanhecke PPE-DE: Albertini, Andrikienė, Antoniozzi, Audy, Ayuso González, Bachelot-Narquin, Barsi-Pataky, Bauer, Becsey, Belet, Berend, Bowis, Brejc, Brepoels, Březina, Brok, Busuttil, Buzek, Carollo, Casa, Caspary, Castiglione, del Castillo Vera, Cesa, Chmielewski, Cirino Pomicino, Coelho, Coveney, Daul, Dehaene, Demetriou, Descamps, Deß, De Veyrac, Díaz de Mera García Consuegra, Dimitrakopoulos, Dionisi, Dombrovskis, Doorn, Doyle, Duka-Zólyomi, Ebner, Esteves, Eurlings, Fatuzzo, Ferber, Fernández Martín, Florenz, Fontaine,