Monogenic Autoinflammatory Diseases with Mendelian Inheritance: Genes, Mutations, and Genotype/Phenotype Correlations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Benefits of Exome Sequencing in Children with Suspected Isolated
G C A T T A C G G C A T genes Article Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss Roxane Van Heurck 1, Maria Teresa Carminho-Rodrigues 1, Emmanuelle Ranza 1, Caterina Stafuzza 2, Lina Quteineh 1, Corinne Gehrig 1, Eva Hammar 1, Michel Guipponi 1, Marc Abramowicz 1, Pascal Senn 2 , Nils Guinand 2, Helene Cao-Van 2 and Ariane Paoloni-Giacobino 1,* 1 Division of Genetic Medicine, Geneva University Hospitals, 1205 Geneva, Switzerland; [email protected] (R.V.H.); [email protected] (M.T.C.-R.); [email protected] (E.R.); [email protected] (L.Q.); [email protected] (C.G.); [email protected] (E.H.); [email protected] (M.G.); [email protected] (M.A.) 2 Ear-Nose-Throat/Head and Neck Surgery Division, Geneva University Hospitals, 1205 Geneva, Switzerland; [email protected] (C.S.); [email protected] (P.S.); [email protected] (N.G.); [email protected] (H.C.-V.) * Correspondence: [email protected] Abstract: Purpose: Hearing loss is characterized by an extensive genetic heterogeneity and remains a common disorder in children. Molecular diagnosis is of particular benefit in children, and permits the early identification of clinically-unrecognized hearing loss syndromes, which permits effective clinical management and follow-up, including genetic counselling. Methods: We performed whole-exome Citation: Van Heurck, R.; sequencing with the analysis of a panel of 189 genes associated with hearing loss in a prospective Carminho-Rodrigues, M.T.; Ranza, E.; cohort of 61 children and 9 adults presenting mainly with isolated hearing loss. -

Dissertationes Medicinae Universitatis Tartuensis 178
DISSERTATIONES MEDICINAE UNIVERSITATIS TARTUENSIS 178 DISSERTATIONES MEDICINAE UNIVERSITATIS TARTUENSIS 178 RITA TEEK The genetic causes of early onset hearing loss in Estonian children Department of Paediatrics, University of Tartu, Tartu, Estonia Dissertation is accepted for commencement of the degree of Doctor of Medical Sciences on September 22, 2010 by the Council of the Faculty of Medicine, University of Tartu, Estonia. Supervisors: Professor Katrin Õunap, MD, PhD, Department of Paediatrics, University of Tartu, Tartu, Estonia The Late Professor Mart Kull, MD, PhD, Department of Oto-Rhino-Laryngology, University of Tartu, Tartu, Estonia (2005–2008) Reviewers: Assistant Professor Gunnar Tasa, MD, PhD, Department of General and Molecular Pathology, University of Tartu, Tartu, Estonia Assistant Professor Oivi Uibo, MD, PhD, Department of Paediatrics, University of Tartu, Tartu, Estonia Opponent: Professor Lisbeth Tranebjærg, MD, PhD, Department of Audiology, H:S Bispebjerg Hospital, and Wilhelm Johannsen Centre of Functional Genomics Institute of Cellular and Molecular Medicine, ICMM, University of Copenhagen, The Panum Institute, Denmark Commencement: November 24, 2010 ISSN 1024–395x ISBN 978–9949–19–478–0 (trükis) ISBN 978–9949–19–479–7 (PDF) Autoriõigus: Rita Teek, 2010 Tartu Ülikooli Kirjastus www.tyk.ee Tellimuse nr. 570 To my patients and their families CONTENTS LIST OF ORIGINAL PUBLICATIONS ...................................................... 9 ABBREVIATIONS OF HEARING LOSS STUDY GROUPS AND PATIENTS .................................................................................................... -

The Pathological Consequences of Impaired Genome Integrity in Humans; Disorders of the DNA Replication Machinery
The pathological consequences of impaired genome integrity in humans; disorders of the DNA replication machinery Article (Accepted Version) O'Driscoll, Mark (2017) The pathological consequences of impaired genome integrity in humans; disorders of the DNA replication machinery. Journal of Pathology, 241 (2). pp. 192-207. ISSN 0022-3417 This version is available from Sussex Research Online: http://sro.sussex.ac.uk/id/eprint/65956/ This document is made available in accordance with publisher policies and may differ from the published version or from the version of record. If you wish to cite this item you are advised to consult the publisher’s version. Please see the URL above for details on accessing the published version. Copyright and reuse: Sussex Research Online is a digital repository of the research output of the University. Copyright and all moral rights to the version of the paper presented here belong to the individual author(s) and/or other copyright owners. To the extent reasonable and practicable, the material made available in SRO has been checked for eligibility before being made available. Copies of full text items generally can be reproduced, displayed or performed and given to third parties in any format or medium for personal research or study, educational, or not-for-profit purposes without prior permission or charge, provided that the authors, title and full bibliographic details are credited, a hyperlink and/or URL is given for the original metadata page and the content is not changed in any way. http://sro.sussex.ac.uk The pathological consequences of impaired genome integrity in humans; disorders of the DNA replication machinery. -

2021 Code Changes Reference Guide
Boston University Medical Group 2021 CPT Code Changes Reference Guide Page 1 of 51 Background Current Procedural Terminology (CPT) was created by the American Medical Association (AMA) in 1966. It is designed to be a means of effective and dependable communication among physicians, patients, and third-party payers. CPT provides a uniform coding scheme that accurately describes medical, surgical, and diagnostic services. CPT is used for public and private reimbursement systems; development of guidelines for medical care review; as a basis for local, regional, and national utilization comparisons; and medical education and research. CPT Category I codes describe procedures and services that are consistent with contemporary medical practice. Category I codes are five-digit numeric codes. CPT Category II codes facilitate data collection for certain services and test results that contribute to positive health outcomes and quality patient care. These codes are optional and used for performance management. They are alphanumeric five-digit codes with the alpha character F in the last position. CPT Category III codes represent emerging technologies. They are alphanumeric five-digit codes with the alpha character T in the last position. The CPT Editorial Panel, appointed by the AMA Board of Trustees, is responsible for maintaining and updating the CPT code set. Purpose The AMA makes annual updates to the CPT code set, effective January 1. These updates include deleted codes, revised codes, and new codes. It’s important for providers to understand the code changes and the impact those changes will have to systems, workflow, reimbursement, and RVUs. This document is meant to assist you with this by providing a summary of the changes; a detailed breakdown of this year’s CPT changes by specialty, and HCPCS Updates for your reference. -

Using Drosophila to Study Mechanisms of Hereditary Hearing Loss Tongchao Li1,*, Hugo J
© 2018. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2018) 11, dmm031492. doi:10.1242/dmm.031492 REVIEW Using Drosophila to study mechanisms of hereditary hearing loss Tongchao Li1,*, Hugo J. Bellen1,2,3,4,5 and Andrew K. Groves1,3,5,‡ ABSTRACT Keats and Corey, 1999; Kimberling et al., 2010; Mathur and Yang, Johnston’s organ – the hearing organ of Drosophila – has a very 2015). It is an autosomal recessive genetic disease, characterized by different structure and morphology to that of the hearing organs of varying degrees of deafness and retinitis pigmentosa-induced vision vertebrates. Nevertheless, it is becoming clear that vertebrate and loss. Although our understanding of genetic hearing loss has invertebrate auditory organs share many physiological, molecular advanced greatly over the past 20 years (Vona et al., 2015), there is a and genetic similarities. Here, we compare the molecular and cellular pressing need for experimental systems to understand the function features of hearing organs in Drosophila with those of vertebrates, of the proteins encoded by deafness genes. The mouse is well and discuss recent evidence concerning the functional conservation established as a model for studying human genetic deafness (Brown of Usher proteins between flies and mammals. Mutations in Usher et al., 2008), but other model organisms, such as the fruit fly genes cause Usher syndrome, the leading cause of human deafness Drosophila, might also provide convenient and more rapid ways to and blindness. In Drosophila, some Usher syndrome proteins appear assay the function of candidate deafness genes. to physically interact in protein complexes that are similar to those In mammals, mechanosensitive hair cells reside in a specialized described in mammals. -

Role of Transfer RNA Modification and Aminoacylation in the Etiology of Congenital Intellectual Disability
Franz et al. J Transl Genet Genom 2020;4:50-70 Journal of Translational DOI: 10.20517/jtgg.2020.13 Genetics and Genomics Review Open Access Role of transfer RNA modification and aminoacylation in the etiology of congenital intellectual disability Martin Franz#, Lisa Hagenau#, Lars R. Jensen, Andreas W. Kuss Department of Functional Genomics, Interfaculty Institute for Genetics and Functional Genomics, University Medicine Greifswald, Greifswald 17475, Germany. #Authors cotributed equally. Correspondence to: Prof. Andreas W. Kuss; Dr. Lars R. Jensen, Department of Functional Genomics, University Medicine Greifswald, C_FunGene, Felix-Hausdorff-Str. 8, Greifswald 17475, Germany. E-mail: [email protected]; [email protected] How to cite this article: Franz M, Hagenau L, Jensen LR, Kuss AW. Role of transfer RNA modification and aminoacylation in the etiology of congenital intellectual disability. J Transl Genet Genom 2020;4:50-70. http://dx.doi.org/10.20517/jtgg.2020.13 Received: 14 Feb 2020 First Decision: 17 Mar 2020 Revised: 30 Mar 2020 Accepted: 23 Apr 2020 Available online: 16 May 2020 Science Editor: Tjitske Kleefstra Copy Editor: Jing-Wen Zhang Production Editor: Tian Zhang Abstract Transfer RNA (tRNA) modification and aminoacylation are post-transcriptional processes that play a crucial role in the function of tRNA and thus represent critical steps in gene expression. Knowledge of the exact processes and effects of the defects in various tRNAs remains incomplete, but a rapidly increasing number of publications over the last decade has shown a growing amount of evidence as to the importance of tRNAs for normal human development, including brain formation and the development and maintenance of higher cognitive functions as well. -

Case Report 12Q14 Microdeletions: Additional Case Series with Confirmation of a Macrocephaly Region
Hindawi Publishing Corporation Case Reports in Genetics Volume 2015, Article ID 192071, 7 pages http://dx.doi.org/10.1155/2015/192071 Case Report 12q14 Microdeletions: Additional Case Series with Confirmation of a Macrocephaly Region Adrian Mc Cormack,1 Cynthia Sharpe,2 Nerine Gregersen,3 Warwick Smith,4 Ian Hayes,3 Alice M. George,1 and Donald R. Love1 1 Diagnostic Genetics, LabPLUS, Auckland City Hospital, P.O. Box 110031, Auckland 1148, New Zealand 2Department of Neuroservices, Starship Children’s Health, Private Bag 92024, Auckland 1142, New Zealand 3Genetic Health Service New Zealand-Northern Hub, Auckland City Hospital, Private Bag 92024, Auckland 1142, New Zealand 4Middlemore Hospital, Private Bag 93311, Otahuhu, Auckland 1640, New Zealand Correspondence should be addressed to Donald R. Love; [email protected] Received 16 April 2015; Revised 7 July 2015; Accepted 7 July 2015 Academic Editor: Patrick Morrison Copyright © 2015 Adrian Mc Cormack et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. To date, there have been only a few reports of patients carrying a microdeletion in chromosome 12q14. These patients usually present with pre- and postnatal growth retardation, and developmental delay. Here we report on two additional patients with both genotype and phenotype differences. Similar to previously published cases, one patient has haploinsufficiency ofthe HMGA2 gene and shows severe short stature and developmental delay. The second patient is only one of a handful without the loss of the HMGA2 gene and shows a much better growth profile, but with absolute macrocephaly. -

A Mouse Model for Nonsyndromic Deafness (DFNB12) Links Hearing Loss to Defects in Tip Links of Mechanosensory Hair Cells
A mouse model for nonsyndromic deafness (DFNB12) links hearing loss to defects in tip links of mechanosensory hair cells Martin Schwandera,b, Wei Xionga,b, Joshua Tokitac, Andrea Lellid, Heather M. Elledgea,b, Piotr Kazmierczaka,b, Anna Sczanieckaa,b, Anand Kolatkara, Tim Wiltshiree,1, Peter Kuhna, Jeffrey R. Holtd, Bechara Kacharc, Lisa Tarantinoe,1, and Ulrich Mu¨ llera,b,2 aDepartment of Cell Biology and bInstitute of Childhood and Neglected Disease, The Scripps Research Institute, La Jolla, CA 92037; cLaboratory of Cellular Biology, National Institute of Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD 20892; dDepartment of Neuroscience, University of Virginia School of Medicine, Charlottesville, VA 22908; and eGenomics Institute of the Novartis Research Foundation, San Diego, CA 92121 Communicated by Bruce A. Beutler, The Scripps Research Institute, La Jolla, CA, January 26, 2009 (received for review January 9, 2009) Deafness is the most common form of sensory impairment in mechanism by which such mutations cause disease is unclear. humans and is frequently caused by single gene mutations. Inter- In an N-ethyl-N-nitrosourea (ENU) mutagenesis screen (35), estingly, different mutations in a gene can cause syndromic and we have now identified salsa mice, which suffer from progres- nonsyndromic forms of deafness, as well as progressive and sive hearing loss and carry a Cdh23 missense mutation that is age-related hearing loss. We provide here an explanation for the predicted to affect Ca2ϩ binding by the extracellular CDH23 phenotypic variability associated with mutations in the cadherin 23 domain. Similar mutations in the human CDH23 gene cause gene (CDH23). -

A Comprehensive Review on Inherited Sensorineural Hearing Loss and Their Syndromes
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 14 August 2020 doi:10.20944/preprints202008.0308.v1 Manuscript (Review Article) A comprehensive review on inherited Sensorineural Hearing Loss and their syndromes Authors Muhammad Noman 1, Shazia Anwer Bukhari 1, Muhammad Tahir 2, & Shehbaz Ali 3* Author’s information 1 Department of Biochemistry, Molecular Biology laboratory, Government College, University, Faisalabad, 38000, Pakistan. 2 Department of Oncology, Allied Teaching Hospital Faisalabad, Faisalabad Medical University, Faisalabad, 38000, Pakistan. 3 Department of Biosciences and Technology, Khwaja Fareed University of Engineering and information technology, Rahim Yar Khan, Punjab, Pakistan. *Correspondence: Shehbaz Ali: [email protected] Tel: +92-333-7477407 Abstract Hearing impairment is an immensely diagnosed genetic cause, 5% of the total world population effects with different kind of congenital hearing loss (HL). In third-world countries or countries where consanguineous marriages are more common the frequency rate of genetic disorders are at its zenith. Approximately, the incidence of hearing afflictions is ostensibly 7-8:1000 individuals whereas it is estimated that about 466 million peoples suffer with significant HL, and of theses deaf cases 34 million are children’s up to March, 2020. Several genes and colossal numbers of pathogenic variants cause hearing impairment, which aided in next-generation with recessive, dominant or X-linked inheritance traits. This review highlights on syndromic and non-syndromic HL (SHL and NSHL), and categorized as conductive, sensorineural and mixed HL, which having autosomal dominant and recessive, and X-linked or mitochondrial mode of inheritance. Many hundred genes involved in HL are reported, and their mutation spectrum becomes very wide. -

WO 2015/048577 A2 April 2015 (02.04.2015) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/048577 A2 April 2015 (02.04.2015) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 48/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 14/057905 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 26 September 2014 (26.09.2014) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/883,925 27 September 2013 (27.09.2013) US (84) Designated States (unless otherwise indicated, for every 61/898,043 31 October 2013 (3 1. 10.2013) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (71) Applicant: EDITAS MEDICINE, INC. -

Autosomal Dominantly Inherited GREB1L Variants in Individuals with Profound Sensorineural Hearing Impairment
G C A T T A C G G C A T genes Article Autosomal Dominantly Inherited GREB1L Variants in Individuals with Profound Sensorineural Hearing Impairment Isabelle Schrauwen 1,* , Khurram Liaqat 2, Isabelle Schatteman 3 , Thashi Bharadwaj 1, Abdul Nasir 4 , Anushree Acharya 1, Wasim Ahmad 5, Guy Van Camp 6 and Suzanne M. Leal 1 1 Center for Statistical Genetics, Sergievsky Center, Taub Institute for Alzheimer’s Disease and the Aging Brain, and the Department of Neurology, Columbia University Medical Center, New York, NY 10032, USA; [email protected] (T.B.); [email protected] (A.A.); [email protected] (S.M.L.) 2 Department of Biotechnology, Faculty of Biological Sciences, Quaid-i-Azam University, Islamabad 45320, Pakistan; [email protected] 3 Department of ENT, St-Augustinus Hospital Antwerp, 2610 Antwerp, Belgium; [email protected] 4 Synthetic Protein Engineering Lab (SPEL), Department of Molecular Science and Technology, Ajou University, Suwon 443-749, Korea; [email protected] 5 Department of Biochemistry, Faculty of Biological Sciences, Quaid-i-Azam University, Islamabad 45320, Pakistan; [email protected] 6 Center of Medical Genetics, University of Antwerp & Antwerp University Hospital, 2650 Antwerp, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +1-(212)-304-5272 Received: 1 June 2020; Accepted: 20 June 2020; Published: 23 June 2020 Abstract: Congenital hearing impairment is a sensory disorder that is genetically highly heterogeneous. By performing exome sequencing in two families with congenital nonsyndromic profound sensorineural hearing loss (SNHL), we identified autosomal dominantly inherited missense variants [p.(Asn283Ser); p.(Thr116Ile)] in GREB1L, a neural crest regulatory molecule. -
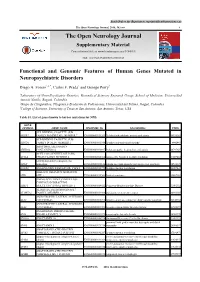
The Open Neurology Journal, 2016, 10, I-Xv I the Open Neurology Journal Supplementary Material Content List Available At
Send Orders for Reprints to [email protected] The Open Neurology Journal, 2016, 10, i-xv i The Open Neurology Journal Supplementary Material Content list available at: www.benthamopen.com/TONEUJ/ DOI: 10.2174/1874205X01610010143 Functional and Genomic Features of Human Genes Mutated in Neuropsychiatric Disorders Diego A. Forero1,4, *, Carlos F. Prada2 and George Perry3 1Laboratory of NeuroPsychiatric Genetics, Biomedical Sciences Research Group, School of Medicine, Universidad Antonio Nariño. Bogotá, Colombia 2Grupo de Citogenética, Filogenia y Evolución de Poblaciones, Universidad del Tolima. Ibagué, Colombia 3College of Sciences, University of Texas at San Antonio, San Antonio, Texas, USA Table S1. List of genes known to harbor mutations for NPD. GENE SYMBOL GENE NAME ENSEMBL ID DISORDERS PMID ATP-BINDING CASSETTE, SUB- ABCB7 FAMILY B (MDR/TAP), MEMBER 7 ENSG00000131269 X-linked sideroblastic anemia and ataxia 10196363 ATP-BINDING CASSETTE, SUB- ABCD1 FAMILY D (ALD), MEMBER 1 ENSG00000101986 X-linked adrenoleukodystrophy 8441467 ABHYDROLASE DOMAIN ABHD12 CONTAINING 12 ENSG00000100997 Polyneuropathy, hearing loss and ataxia 20797687 ACYL-COA SYNTHETASE LONG- ACSL4 CHAIN FAMILY MEMBER 4 ENSG00000068366 nonspecific X-linked mental retardation 11889465 activity-dependent neuroprotector ADNP homeobox ENSG00000101126 autism spectrum disorder and intellectual disability 24531329 AGTR2 ANGIOTENSIN II RECEPTOR, TYPE 2 ENSG00000180772 X-linked mental retardation 12089445 ABELSON HELPER INTEGRATION AHI1 SITE 1 ENSG00000135541 Joubert