Toll-Like Receptor Signalling Pathways Regulate Hypoxic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amphioxus TLR Signaling Adaptors Act As Negative Regulators In
Novel Toll/IL-1 Receptor Homologous Region Adaptors Act as Negative Regulators in Amphioxus TLR Signaling This information is current as Jian Peng, Xin Tao, Rui Li, Jingru Hu, Jie Ruan, Ruihua of September 29, 2021. Wang, Manyi Yang, Rirong Yang, Xiangru Dong, Shangwu Chen, Anlong Xu and Shaochun Yuan J Immunol published online 31 August 2015 http://www.jimmunol.org/content/early/2015/08/30/jimmun ol.1403003 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2015/08/30/jimmunol.140300 Material 3.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 29, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published August 31, 2015, doi:10.4049/jimmunol.1403003 The Journal of Immunology Novel Toll/IL-1 Receptor Homologous Region Adaptors Act as Negative Regulators in Amphioxus TLR Signaling Jian Peng,*,1 Xin Tao,* Rui Li,* Jingru Hu,* Jie Ruan,* Ruihua Wang,* Manyi Yang,* Rirong Yang,* Xiangru Dong,* Shangwu Chen,* Anlong Xu,*,† and Shaochun Yuan* Studies have shown that the basal chordate amphioxus possesses an extraordinarily complex TLR system, including 39 TLRs and at least 40 Toll/IL-1R homologous region (TIR) adaptors. -

A Truncated TIR-NBS Protein TN10 Pairs with Two Clustered TIR-NBS-LRR Immune Receptors and Contributes to Plant Immunity in Arabidopsis
International Journal of Molecular Sciences Article A Truncated TIR-NBS Protein TN10 Pairs with Two Clustered TIR-NBS-LRR Immune Receptors and Contributes to Plant Immunity in Arabidopsis Yongming Chen 1, Guitao Zhong 1,2, Huiren Cai 1,2, Renjie Chen 1, Na Liu 1, Wei Wang 1,* and Dingzhong Tang 1,* 1 State Key Laboratory of Ecological Control of Fujian-Taiwan Crop Pests, Key Laboratory of Ministry of Education for Genetics, Breeding and Multiple Utilization of Crops, Plant Immunity Center, Fujian Agriculture and Forestry University, Fuzhou 350002, China; [email protected] (Y.C.); [email protected] (G.Z.); [email protected] (H.C.); [email protected] (R.C.); [email protected] (N.L.) 2 College of Life Science, Fujian Agriculture and Forestry University, Fuzhou 350002, China * Correspondence: [email protected] (W.W.); [email protected] (D.T.) Abstract: The encoding genes of plant intracellular nucleotide-binding site (NBS) and leucine-rich repeat (LRR) domain receptors (NLRs) often exist in the form of a gene cluster. Several recent studies demonstrated that the truncated Toll/interleukin-1 receptor-NBS (TIR-NBS) proteins play important roles in immunity. In this study, we identified a large TN gene cluster on Arabidopsis ecotype Col-0 chromosome 1, which included nine TN genes, TN4 to TN12. Interestingly, this cluster also contained two typical TIR-NBS-LRR genes: At1g72840 and At1g72860 (hereinafter referred to as TNL40 and Citation: Chen, Y.; Zhong, G.; TNL60, respectively), which formed head-to-head genomic arrangement with TN4 to TN12. However, Cai, H.; Chen, R.; Liu, N.; Wang, W.; the functions of these TN and TNL genes in this cluster are still unknown. -

Enteropathogenic Escherichia Coli Tir Recruits Cellular SHP-2 Through ITIM Motifs to Suppress Host Immune Response☆
Cellular Signalling 25 (2013) 1887–1894 Contents lists available at SciVerse ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig Enteropathogenic Escherichia coli Tir recruits cellular SHP-2 through ITIM motifs to suppress host immune response☆ Dapeng Yan a,1, Heming Quan b,1, Lin Wang b, Feng Liu a, Haipeng Liu b, Jianxia Chen b, Xuetao Cao c, Baoxue Ge a,b,⁎ a Institute of Health Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences & Shanghai Jiaotong University School of Medicine, Shanghai, China b Shanghai TB Key Laboratory, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, China c National Key Laboratory of Medical Immunology and Institute of Immunology, Second Military Medical University, Shanghai, China article info abstract Article history: Immune responses to pathogens are regulated by immune receptors containing either an immunoreceptor Received 28 January 2013 tyrosine-based activation motif (ITAM) or an immunoreceptor tyrosine-based inhibitory motif (ITIM). Received in revised form 9 May 2013 The important diarrheal pathogen enteropathogenic Escherichia coli (EPEC) require delivery and insertion Accepted 12 May 2013 of the bacterial translocated intimin receptor (Tir) into the host plasma membrane for pedestal formation. Available online 23 May 2013 The C-terminal region of Tir, encompassing Y483 and Y511, shares sequence similarity with cellular ITIMs. fl Keywords: Here, we show that EPEC Tir suppresses the production of in ammatory cytokines by recruitment of SHP-2 fi Enteropathogenic Escherichia coli and subsequent deubiquitination of TRAF6 in an ITIM dependent manner. Our ndings revealed a novel Tir mechanism by which the EPEC utilize its ITIM motifs to suppress and evade the host innate immune re- SHP-2 sponse, which could lead to the development of novel therapeutics to prevent bacterial infection. -

Z7g01110005920.Pdf
Retina Effects of Ischemic Preconditioning and Bevacizumab on Apoptosis and Vascular Permeability Following Retinal Ischemia–Reperfusion Injury Steven F. Abcouwer,1,2,3 Cheng-mao Lin,2 Ellen B. Wolpert,2 Sumathi Shanmugam,2 Eric W. Schaefer,4 Willard M. Freeman,5 Alistair J. Barber,3 and David A. Antonetti2,3 PURPOSE. Using transient ischemia followed by reperfusion (IR) etinal neovascularization and macular edema are associ- to model ischemic retinal disease, this study compares the Rated with vision loss in several retinal diseases, including effects of ischemic preconditioning (IPC) and therapies target- age-related macular degeneration (AMD) and ischemic retinop- ing vascular endothelial growth factor (VEGF) and tumor ne- athies, such as retinal vascular occlusive diseases and diabetic crosis factor (TNF)-␣ on retinal apoptosis, vascular permeabil- retinopathy. Although the vascular components are most ob- ity, and mRNA expression. vious, these retinopathies are also associated with various de- METHODS. Rats were subjected to 30 or 45 minutes of retinal grees of inflammation and neurodegeneration. For example, development of AMD is linked to complement activation and ischemia followed by reperfusion for up to 48 hours. Neuro- 1,2 degeneration was quantified by caspase-3 (DEVDase) activity inflammation. Diabetic retinopathy is associated with micro- and by measuring nucleosomal DNA content (cell death glial activation, increased expression of inflammatory cyto- kines, adherence of leukocytes to the retinal microvasculature ELISA). Vascular leakage was quantified by the Evans Blue dye 3–6 method. A set of IR-responsive mRNAs was identified by whole- (leukostasis), and apoptotic death of neurons. The func- genome microarray and confirmed by RT-PCR analyses. -

Antiviral Activity of Bacterial TIR Domains Via Signaling Molecules That Trigger Cell Death
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.06.425286; this version posted January 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Antiviral activity of bacterial TIR domains via signaling molecules that trigger cell death Gal Ofir1, Ehud Herbst1, Maya Baroz1, Daniel Cohen1, Adi Millman1, Shany Doron1, Nitzan Tal1, Daniel B. A. Malheiro2, Sergey Malitsky3, Gil Amitai1,#, Rotem Sorek1,# 1 Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel 2 MS-Omics, DK-2950 Vedbæk, Denmark 3 Life Science Core Facilities, Weizmann Institute of Science, Rehovot, Israel. # Correspondence: [email protected] , [email protected] Abstract The Toll/interleukin-1 receptor (TIR) domain is a canonical component of animal and plant immune systems. In plants, intracellular pathogen sensing by immune receptors triggers their TIR domains to generate a molecule which is a variant of cyclic ADP-ribose (v-cADPR). This molecule is hypothesized to activate plant cell death via a yet unresolved pathway. TIR domains were recently also shown to be involved in a bacterial anti-phage defense system called Thoeris, but the mechanism of Thoeris defense remained unknown. In this study we report that phage infection triggers Thoeris TIR-domain proteins to produce an isomer of cyclic ADP-ribose. This molecular signal activates a second protein, ThsA, which then depletes the cell of the essential molecule nicotinamide adenine dinucleotide (NAD) and leads to abortive infection and cell death. -

CEACAM1 Regulates the IL-6 Mediated Fever
Zhang et al. BMC Immunology (2019) 20:7 https://doi.org/10.1186/s12865-019-0287-y RESEARCH ARTICLE Open Access CEACAM1 regulates the IL-6 mediated fever response to LPS through the RP105 receptor in murine monocytes Zhifang Zhang*, Deirdre La Placa, Tung Nguyen, Maciej Kujawski, Keith Le, Lin Li and John E. Shively* Abstract Background: Systemic inflammation and the fever response to pathogens are coordinately regulated by IL-6 and IL-1β. We previously showed that CEACAM1 regulates the LPS driven expression of IL-1β in murine neutrophils through its ITIM receptor. Results: We now show that the prompt secretion of IL-6 in response to LPS is regulated by CEACAM1 expression on bone marrow monocytes. Ceacam1−/− mice over-produce IL-6 in response to an i.p. LPS challenge, resulting in prolonged surface temperature depression and overt diarrhea compared to their wild type counterparts. Intraperitoneal injection of a 64Cu-labeled LPS, PET imaging agent shows confined localization to the peritoneal cavity, and fluorescent labeled LPS is taken up by myeloid splenocytes and muscle endothelial cells. While bone marrow monocytes and their progenitors (CD11b+Ly6G−) express IL-6 in the early response (< 2 h) to LPS in vitro, these cells are not detected in the bone marrow after in vivo LPS treatment perhaps due to their rapid and complete mobilization to the periphery. Notably, tissue macrophages are not involved in the early IL-6 response to LPS. In contrast to human monocytes, TLR4 is not expressed on murine bone marrow monocytes. Instead, the alternative LPS receptor RP105 is expressed and recruits MD1, CD14, Src, VAV1 and β-actin in response to LPS. -

Interaction of Urokinase Receptor and Integrins on Endothelial Cells and Its Implication in VEGF-Induced Migration
Interaction of urokinase receptor and integrins on endothelial cells and its implication in VEGF-induced migration Doctoral thesis at the Medical University of Vienna In partial fulfillment for the degree of Doctor of Philosophy (PhD) Submitted by Revu Ann Alexander Supervisor: Univ.Prof.Dr.Michael Freissmuth,MD Institute for Vascular Biology and Thrombosis Research Center for Physiology and Pharmacology Medical University of Vienna Schwarzpanierstrasse 17 A-1090 Vienna, Austria. Vienna, January 2012 ACKNOWLEDGEMENTS As I now stand at the precipice of this great odyssey, which is the PhD, I do agree. Science is not for the faint-hearted. One day you are at the top of the world brimming with exciting results and ideas, and the next day everything can come crashing down. But the entire experience was not only about learning scientific techniques, rational questioning and thinking out of the box. It was also about learning to believe in myself, to be strong and confident. And for that I am humbly grateful to many. To late Prof.Bernd.B.Binder, who gave me the opportunity to pursue my interest in science. For giving “carte blanche” to try out ideas, and encouraging me to look at the bright side no matter what the outcome was. I hope I can emulate your spirit and passion for life and science. To Prof.Michael Freissmuth, for the selflessness and generosity shown at difficult times. For his relentless enthusiasm to encourage students to think, ask and prode science. Hopefully I have imbibed some of his lessons. To Prof. Johannes.M.Breuss, for the discussions scientific and otherwise; and especially the willingness to help out with any difficulties. -
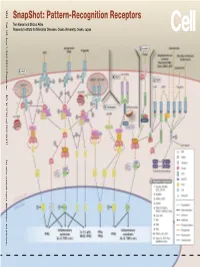
Snapshot: Pattern-Recognition Receptors
SnapShot: Pattern-Recognition Receptors SnapShot:Pattern-Recognition Kawai and Shizuo Akira Taro Osaka, Japan Diseases, Osaka University, Institute for Microbial Research 1024 Cell 129, June 1, 2007 ©2007 Elsevier Inc. DOI 10.1016/j.cell.2007.05.017 See online version for legend, abbreviations, and references. SnapShot: Pattern-Recognition Receptors Taro Kawai and Shizuo Akira Research Institute for Microbial Diseases, Osaka University, Osaka, Japan (A) Toll-like receptor signaling. Toll-like receptor (TLR) 3 recognizes polyinosinic-polycytidylic acid (poly IC), whereas TLR4 recognizes lipopolysaccharide (LPS). TLR2 recog- nizes various components such as lipoprotein and peptidoglycan (PGN). TLR5 detects flagellin. TLR7 and TLR9 detect single-stranded (ss)RNA and CpG DNA, respectively. Each TLR recruits a distinct set of Toll/interleukin-1 receptor (TIR) domain-containing adaptor molecules such as myeloid differentiation primary response gene 88 (MyD88), TIR-containing adaptor protein (TIRAP, also known as MAL), TIR-containing adaptor-inducing IFNβ (TRIF, also known as TICAM1) and TRIF-related adaptor molecule (TRAM, also known as TICAM2). TLR3 uses TRIF, and TLR5, 7, and 9 use MyD88. TLR2 uses MyD88 and TIRAP, and TLR4 uses MyD88, TIRAP, TRIF, and TRAM. MyD88 binds to inter- leukin-1 receptor-associated kinase 4 (IRAK4) and TRAF6. TRIF binds receptor-interacting protein 1 (RIP1) and TRAF6. TRAF6 forms a complex with Ubc13, Uev1A, and ECSIT (evolutionarily conserved signaling intermediate in Toll/IL-1R pathways) to activate a complex containing transforming growth factor-β-activated kinase 1 (TAK1), TAK1-binding protein 1 (TAB1), TAB2, and TAB3. TAK1 activates IκB kinase (IKK) complex consisting of IKKα, IKKβ, and Nemo (also known as IKKγ), which results in the phosphorylation and proteasomal degradation of IκB proteins and the release of a transcription factor NFκB to the nucleus to regulate expression of inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor α (TNFα). -

SCIMP Is a Universal Toll‐Like Receptor Adaptor in Macrophages
SCIMP is a universal Toll-like receptor adaptor controlling selective cytokine outputs from macrophages Lin Luo*, James E. B. Curson, Liping Liu, Adam A. Wall, Neeraj Tuladhar, Richard M. Lucas, Matthew J. Sweet and Jennifer L. Stow Institute for Molecular Bioscience (IMB) and IMB Centre for Inflammation and Disease Research, The University of Queensland, Brisbane, QLD 4072, Australia Summary sentence: The TLR adaptor SCIMP is used by multiple TLRs to generate cytokine specificity in macrophages *Correspondence should be addressed to: Dr Lin Luo Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia. Email: [email protected] Key words: SCIMP, Toll-like receptor, TRAP, inflammatory cytokines, macrophage, effectors This is the author manuscript accepted for publication and has undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1002/JLB.2MA0819-138RR. This article is protected by copyright. All rights reserved. Word counts: 4721 This article is protected by copyright. All rights reserved. ABSTRACT In innate immune cells, pathogens and danger signals activate Toll-like receptors (TLRs), unleashing potent and tailored inflammatory responses. Previously, we reported that an immune-specific transmembrane adaptor, SCIMP, interacts with TLR4 via direct binding to its cytoplasmic TIR domain. SCIMP scaffolds a Src family kinase, Lyn, for TLR4 phosphorylation and activation. Consequently, SCIMP is able to direct selective production of the pro-inflammatory cytokines IL-6 and IL-12p40 downstream of TLR4 in macrophages. Here we set out to investigate whether SCIMP also acts as an adaptor for other TLR family members. -

Rab6-Dependent Retrograde Traffic of LAT Controls Immune Synapse Formation and T Cell Activation
Rab6-dependent retrograde traffic of LAT controls immune synapse formation and T cell activation Jean-Marie Carpier, Andres Zucchetti, Laurence Bataille, Stephanie Dogniaux, Massiullah Shafaq-Zadah, Sabine Bardin, Marco Lucchino, Mathieu Maurin, Leonel Joannas, Joao Gamelas Magalhaes, et al. To cite this version: Jean-Marie Carpier, Andres Zucchetti, Laurence Bataille, Stephanie Dogniaux, Massiullah Shafaq- Zadah, et al.. Rab6-dependent retrograde traffic of LAT controls immune synapse formation and T cell activation. Journal of Experimental Medicine, Rockefeller University Press, 2018, 215 (4), pp.1245-1265. 10.1084/jem.20162042. inserm-02446749 HAL Id: inserm-02446749 https://www.hal.inserm.fr/inserm-02446749 Submitted on 21 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Article Rab6-dependent retrograde traffic of LAT controls immune synapse formation and T cell activation Jean-Marie Carpier,1* Andres E. Zucchetti,1* Laurence Bataille,1** Stéphanie Dogniaux,1** Massiullah Shafaq-Zadah,3** Sabine Bardin,2 Marco Lucchino,3 Mathieu Maurin,1 -

Regulation of Inflammation and Autophagy in Cancer Biology
Thomas Jefferson University Jefferson Digital Commons Department of Pathology, Anatomy, and Cell Department of Pathology, Anatomy, and Cell Biology Faculty Papers Biology 1-1-2017 Proteoglycan neofunctions: regulation of inflammation and autophagy in cancer biology. Liliana Schaefer Klinikum der Goethe-Universitï Claudia Tredup Klinikum der Goethe-Universitï Maria A. Gubbiotti Thomas Jefferson University Renato V. Iozzo Thomas Jefferson University Follow this and additional works at: https://jdc.jefferson.edu/pacbfp Part of the Oncology Commons, and the Pathology Commons Let us know how access to this document benefits ouy Recommended Citation Schaefer, Liliana; Tredup, Claudia; Gubbiotti, Maria A.; and Iozzo, Renato V., "Proteoglycan neofunctions: regulation of inflammation and autophagy in cancer biology." (2017). Department of Pathology, Anatomy, and Cell Biology Faculty Papers. Paper 230. https://jdc.jefferson.edu/pacbfp/230 This Article is brought to you for free and open access by the Jefferson Digital Commons. The Jefferson Digital Commons is a service of Thomas Jefferson University's Center for Teaching and Learning (CTL). The Commons is a showcase for Jefferson books and journals, peer-reviewed scholarly publications, unique historical collections from the University archives, and teaching tools. The Jefferson Digital Commons allows researchers and interested readers anywhere in the world to learn about and keep up to date with Jefferson scholarship. This article has been accepted for inclusion in Department of Pathology, Anatomy, and Cell Biology Faculty Papers by an authorized administrator of the Jefferson Digital Commons. For more information, please contact: [email protected]. HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author FEBS J Manuscript. Author Author manuscript; Manuscript Author available in PMC 2018 January 01. -

( 12 ) Patent Application Publication ( 10 ) Pub . No .: US 2020/0377591 A1 Majeti Et Al
US 20200377591A1 IN ( 19 ) United States ( 12 ) Patent Application Publication ( 10 ) Pub . No .: US 2020/0377591 A1 Majeti et al . ( 43 ) Pub . Date : Dec. 3 , 2020 ( 54 ) MARKERS OF ACUTE MYELOID ( 60 ) Provisional application No. 61 / 011,324 , filed on Jan. LEUKEMIA STEM CELLS 15 , 2008 . ( 71 ) Applicant: The Board of Trustees of the Leland Publication Classification Stanford Junior University , Stanford , ( 51 ) Int . Ci . CA (US ) COZK 16/28 ( 2006.01 ) C12Q 1/6886 ( 2006.01 ) ( 72 ) Inventors: Ravindra Majeti, Palo Alto , CA ( US ) ; GOIN 33/50 ( 2006.01 ) Irving L. Weissman , Stanford , CA A61P 35/02 ( 2006.01 ) ( US ) GOIN 33/574 ( 2006.01 ) C12N 5/00 ( 2006.01 ) ( 21 ) Appl. No .: 16 / 853,319 C12N 5/09 ( 2006.01 ) CO7K 16/30 ( 2006.01 ) ( 22 ) Filed : Apr. 20 , 2020 ( 52 ) U.S. CI . CPC CO7K 16/2803 ( 2013.01 ) ; CO7K 16/2896 Related U.S. Application Data ( 2013.01 ) ; C12Q 1/6886 ( 2013.01 ) ; GOIN ( 63 ) Continuation of application No. 15 / 704,790 , filed on 33/5011 ( 2013.01 ) ; A61P 35/02 ( 2018.01 ) ; Sep. 14 , 2017 , now Pat . No. 10,662,242 , which is a A61K 2039/505 ( 2013.01 ) ; C12N 570093 continuation of application No. 14 / 927,349 , filed on ( 2013.01 ) ; C12N 5/0694 ( 2013.01 ) ; C07K Oct. 29 , 2015 , now Pat . No. 9,796,781 , which is a 16/28 ( 2013.01 ) ; CO7K 16/30 ( 2013.01 ) ; continuation of application No. 14 / 164,009 , filed on GO1N 33/5 7426 ( 2013.01 ) Jan. 24 , 2014 , now Pat . No. 9,193,955 , which is a ( 57 ) ABSTRACT continuation of application No.