Supporting Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Analysis of DNA Methylation In
Villalba‑Benito et al. Clin Epigenet (2021) 13:51 https://doi.org/10.1186/s13148‑021‑01040‑6 RESEARCH Open Access Genome‑wide analysis of DNA methylation in Hirschsprung enteric precursor cells: unraveling the epigenetic landscape of enteric nervous system development Leticia Villalba‑Benito1,2, Daniel López‑López3,4, Ana Torroglosa1,2, Carlos S. Casimiro‑Soriguer3,4, Berta Luzón‑Toro1,2, Raquel María Fernández1,2, María José Moya‑Jiménez5, Guillermo Antiñolo1,2, Joaquín Dopazo2,3,4 and Salud Borrego1,2* Abstract Background: Hirschsprung disease (HSCR, OMIM 142623) is a rare congenital disorder that results from a failure to fully colonize the gut by enteric precursor cells (EPCs) derived from the neural crest. Such incomplete gut coloniza‑ tion is due to alterations in EPCs proliferation, survival, migration and/or diferentiation during enteric nervous system (ENS) development. This complex process is regulated by a network of signaling pathways that is orchestrated by genetic and epigenetic factors, and therefore alterations at these levels can lead to the onset of neurocristopathies such as HSCR. The goal of this study is to broaden our knowledge of the role of epigenetic mechanisms in the disease context, specifcally in DNA methylation. Therefore, with this aim, a Whole‑Genome Bisulfte Sequencing assay has been performed using EPCs from HSCR patients and human controls. Results: This is the frst study to present a whole genome DNA methylation profle in HSCR and reveal a decrease of global DNA methylation in CpG context in HSCR patients compared with controls, which correlates with a greater hypomethylation of the diferentially methylated regions (DMRs) identifed. -

Pancreatic Beta Cells Express a Diverse Set Ofhomeobox Genes
Proc. Nati. Acad. Sci. USA Vol. 91, pp. 12203-12207, December 1994 Biochemistry Pancreatic beta cells express a diverse set of homeobox genes (Lim motif/Lmx gene/Nkx gene/Alx gene/Vdx homeobox) ABRAHAM RUDNICK*t, THAI YEN LING*, HIROKI ODAGIRI*, WILLIAM J. RUTTER*t, AND MICHAEL S. GERMAN*t§ *Hormone Research Institute and Departments of tMedicine and tBiochemistry and Biophysics, University of California, San Francisco, CA 94143-0534 Contributed by William J. Rutter, August 22, 1994 ABSTRACT Homeobox genes, which are found in all RIPE3B element (16) and the P1 element (8) [also called CT1 eukaryotic organisms, encode transcriptional regulators in- (9)] lie on either side of the IEB1 element. The A/T elements volved in cell-type differentiation and development. Several and the E boxes function synergistically: none of the ele- homeobox genes encoding homeodomain proteins that bind and ments can function in isolation, but combination of an E box activate the insulin gene promoter have been described. In an and an A/T element results in dramatic activation of tran- attempt to identify additional beta-cell homeodomain proteins, scription (11, 16, 19). A number of complexes from beta-cell we designed primers based on the sequences of beta-cell nuclei bind to the A/T elements (6, 8-11, 16, 19). Some homeobox genes cdx3 and lmxl and the Drosophia homeodo- proteins in these complexes have been cloned, and they all main protein Antennapedia and used these primers to amplffy contain homeodomains. The A/T-binding proteins that have inserts by PCR from an insulinoma cDNA library. -

Functional Parsing of Driver Mutations in the Colorectal Cancer Genome Reveals Numerous Suppressors of Anchorage-Independent
Supplementary information Functional parsing of driver mutations in the colorectal cancer genome reveals numerous suppressors of anchorage-independent growth Ugur Eskiocak1, Sang Bum Kim1, Peter Ly1, Andres I. Roig1, Sebastian Biglione1, Kakajan Komurov2, Crystal Cornelius1, Woodring E. Wright1, Michael A. White1, and Jerry W. Shay1. 1Department of Cell Biology, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390-9039. 2Department of Systems Biology, University of Texas M.D. Anderson Cancer Center, Houston, TX 77054. Supplementary Figure S1. K-rasV12 expressing cells are resistant to p53 induced apoptosis. Whole-cell extracts from immortalized K-rasV12 or p53 down regulated HCECs were immunoblotted with p53 and its down-stream effectors after 10 Gy gamma-radiation. ! Supplementary Figure S2. Quantitative validation of selected shRNAs for their ability to enhance soft-agar growth of immortalized shTP53 expressing HCECs. Each bar represents 8 data points (quadruplicates from two separate experiments). Arrows denote shRNAs that failed to enhance anchorage-independent growth in a statistically significant manner. Enhancement for all other shRNAs were significant (two tailed Studentʼs t-test, compared to none, mean ± s.e.m., P<0.05)." ! Supplementary Figure S3. Ability of shRNAs to knockdown expression was demonstrated by A, immunoblotting for K-ras or B-E, Quantitative RT-PCR for ERICH1, PTPRU, SLC22A15 and SLC44A4 48 hours after transfection into 293FT cells. Two out of 23 tested shRNAs did not provide any knockdown. " ! Supplementary Figure S4. shRNAs against A, PTEN and B, NF1 do not enhance soft agar growth in HCECs without oncogenic manipulations (Student!s t-test, compared to none, mean ± s.e.m., ns= non-significant). -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Structural Variant Calling by Assembly in Whole Human Genomes: Applications in Hypoplastic Left Heart Syndrome by Matthew Kendzi
STRUCTURAL VARIANT CALLING BY ASSEMBLY IN WHOLE HUMAN GENOMES: APPLICATIONS IN HYPOPLASTIC LEFT HEART SYNDROME BY MATTHEW KENDZIOR THESIS Submitted in partial FulFillment oF tHe requirements for the degree of Master of Science in BioinFormatics witH a concentration in Crop Sciences in the Graduate College of the University oF Illinois at Urbana-Champaign, 2019 Urbana, Illinois Master’s Committee: ProFessor MattHew Hudson, CHair ResearcH Assistant ProFessor Liudmila Mainzer ProFessor SaurabH SinHa ABSTRACT Variant discovery in medical researcH typically involves alignment oF sHort sequencing reads to the human reference genome. SNPs and small indels (variants less than 50 nucleotides) are the most common types oF variants detected From alignments. Structural variation can be more diFFicult to detect From short-read alignments, and thus many software applications aimed at detecting structural variants From short read alignments have been developed. However, these almost all detect the presence of variation in a sample using expected mate-pair distances From read data, making them unable to determine the precise sequence of the variant genome at the speciFied locus. Also, reads from a structural variant allele migHt not even map to the reference, and will thus be lost during variant discovery From read alignment. A variant calling by assembly approacH was used witH tHe soFtware Cortex-var for variant discovery in Hypoplastic Left Heart Syndrome (HLHS). THis method circumvents many of the limitations oF variants called From a reFerence alignment: unmapped reads will be included in a sample’s assembly, and variants up to thousands of nucleotides can be detected, with the Full sample variant allele sequence predicted. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Mouse Alx3: an Aristaless-Like Homeobox Gene Expressed During Embryogenesis in Ectomesenchyme and Lateral Plate Mesoderm
DEVELOPMENTAL BIOLOGY 199, 11–25 (1998) ARTICLE NO. DB988921 View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Mouse Alx3: An aristaless-like Homeobox Gene Expressed during Embryogenesis in Ectomesenchyme and Lateral Plate Mesoderm Derk ten Berge, Antje Brouwer, Sophia El Bahi,* Jean-Louis Gue´net,† Benoıˆt Robert,* and Frits Meijlink Hubrecht Laboratory, Netherlands Institute for Developmental Biology, Uppsalalaan 8, 3584CT Utrecht, The Netherlands; *Institut Pasteur, De´partement de Biologie Mole´culaireGe´ne´tique Mole´culaire du De´veloppement, 28, rue du Dr. Roux, 75724 Paris Cedex 15, France; and †Institut Pasteur, Ge´ne´tique des Mammife`res, De´partement d’ Immunologie, 28, rue du Dr. Roux, 75724 Paris Cedex 15, France Mouse Alx3 is a homeobox gene that is related to the Drosophila aristaless gene and to a group of vertebrate genes including Prx1, Prx2, Cart1, and Alx4. The protein encoded contains a diverged variant of a conserved peptide sequence present near the carboxyl terminus of at least 15 different paired-class-homeodomain proteins. Alx3 is expressed in mouse embryos from 8 days of gestation onward in a characteristic pattern, predominantly in neural crest-derived mesenchyme and in lateral plate mesoderm. We detected prominent expression in frontonasal head mesenchyme and in the first and second pharyngeal arches and some of their derivatives. High expression was also seen in the tail and in many derivatives of the lateral plate mesoderm including the limbs, the body wall, and the genital tubercle. aristaless-related genes like Alx3, Cart1, and Prx2 are expressed in overlapping proximodistal patterns in the pharyngeal arches. -

Table 3: Average Gene Expression Profiles by Chromosome
Supplemental Data Table 1: Experimental Setup Correlation Array Reverse Fluor Array Extraction Coefficient Print Batch (Y/N) mean (range) DLD1-I.1 I A N DLD1-I.2 I B N 0.86 DLD1-I.3 I C N (0.79-0.90) DLD1-I.4 I C Y DLD1 DLD1-II.1 II D N DLD1-II.2 II E N 0.86 DLD1-II.3 II F N (0.74-0.94) DLD1-II.4 II F Y DLD1+3-II.1 II A N DLD1+3-II.2 II A N 0.85 DLD1 + 3 DLD1+3-II.3 II B N (0.64-0.95) DLD1+3-II.4 II B Y DLD1+7-I.1 I A N DLD1+7-I.2 I A N 0.79 DLD1 + 7 DLD1+7-I.3 I B N (0.68-0.90) DLD1+7-I.4 I B Y DLD1+13-I.1 I A N DLD1+13-I.2 I A N 0.88 DLD1 + 13 DLD1+13-I.3 I B N (0.84-0.91) DLD1+13-I.4 I B Y hTERT-HME-I.1 I A N hTERT-HME-I.2 I B N 0.85 hTERT-HME hTERT-HME-I.3 I C N (0.80-0.92) hTERT-HME-I.4 I C Y hTERT-HME+3-I.1 I A N hTERT-HME+3-I.2 I B N 0.84 hTERT-HME + 3 hTERT-HME+3-I.3 I C N (0.74-0.90) hTERT-HME+3-I.4 I C Y Supplemental Data Table 2: Average gene expression profiles by chromosome arm DLD1 hTERT-HME Ratio.7 Ratio.1 Ratio.3 Ratio.3 Chrom. -

Dynamic Regulatory Module Networks for Inference of Cell Type
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.18.210328; this version posted July 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Dynamic regulatory module networks for inference of cell type specific transcriptional networks Alireza Fotuhi Siahpirani1,2,+, Deborah Chasman1,8,+, Morten Seirup3,4, Sara Knaack1, Rupa Sridharan1,5, Ron Stewart3, James Thomson3,5,6, and Sushmita Roy1,2,7* 1Wisconsin Institute for Discovery, University of Wisconsin-Madison 2Department of Computer Sciences, University of Wisconsin-Madison 3Morgridge Institute for Research 4Molecular and Environmental Toxicology Program, University of Wisconsin-Madison 5Department of Cell and Regenerative Biology, University of Wisconsin-Madison 6Department of Molecular, Cellular, & Developmental Biology, University of California Santa Barbara 7Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison 8Present address: Division of Reproductive Sciences, Department of Obstetrics and Gynecology, University of Wisconsin-Madison +These authors contributed equally. *To whom correspondence should be addressed. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.18.210328; this version posted July 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Changes in transcriptional regulatory networks can significantly alter cell fate. To gain insight into transcriptional dynamics, several studies have profiled transcriptomes and epigenomes at different stages of a developmental process. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Transcriptome Profiling of Cleft Palate Intgf-Beta3 Knockout Mice Alleles: RNA-SEQ Analysis of TGF-Beta3 Mice" (2017)
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Fall 12-15-2017 Transcriptome Profiling of Cleft alateP inTGF-beta3 Knockout Mice Alleles: RNA-SEQ Analysis of TGF-beta3 Mice Kelsey White University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Bioinformatics Commons, and the Developmental Biology Commons Recommended Citation White, Kelsey, "Transcriptome Profiling of Cleft Palate inTGF-beta3 Knockout Mice Alleles: RNA-SEQ Analysis of TGF-beta3 Mice" (2017). Theses & Dissertations. 239. https://digitalcommons.unmc.edu/etd/239 This Thesis is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. TRANSCRIPTOME PROFILING OF CLEFT PALATE IN TGF-3 KNOCKOUT MICE ALLELES: RNA-SEQ ANALYSIS OF TGF-3 MICE By Kelsey Marie White, D.D.S. A THESIS Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of Requirements for the Degree of Master of Science Medical Sciences Interdepartmental Area Graduate Program (Oral Biology) Under the Supervision of Ali Nawshad, Ph.D. University of Nebraska Medical Center Omaha, Nebraska December, 2017 Advisory Committee: Peter Giannini, D.D.S., M.S Ali Nawshad, Ph.D. S. Prem Premaraj, B.D.S., M.S., Ph. D. FRCD(C) Hasan Otu, Ph.D. i ACKNOWLEDGEMENTS First, I would like to sincerely thank my leader and mentor, Dr. Ali Nawshad, for the opportunity to work with and learn from him. -
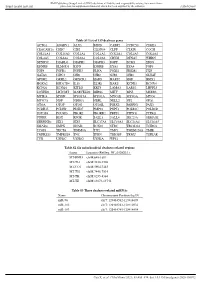
Table S1 List of 159 Deafness Genes ACTG1 ADGRV1 ALX3 BSND
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Med Genet Table S1 List of 159 deafness genes ACTG1 ADGRV1 ALX3 BSND CABP2 CCDC50 CDH23 CEACAM16 CHD7 CIB2 CLDN14 CLPP CLRN1 COCH COL11A1 COL11A2 COL1A1 COL1A2 COL2A1 COL4A3 COL4A4 COL4A5 COL4A6 COL9A1 COL9A2 CRYM DFNA5 WHRN DFNB59 DIABLO DIAPH1 DIAPH3 DSPP ECM1 EDN3 EDNRB ELMOD3 ESPN ESRRB EYA1 EYA4 FGF3 FGF8 FGFR1 FGFR3 FLNA FOXI1 FREM1 FXN GATA3 GIPC3 GJB1 GJB2 GJB3 GJB6 GLYAT GPSM2 GRHL2 GRXCR1 HARS HARS2 HGF HMX1 HOXA2 HSD17B4 IL13 ILDR1 KARS KCNE1 KCNJ10 KCNQ1 KCNQ4 KITLG KRT9 LAMA3 LARS2 LHFPL5 LOXHD1 LRTOMT MARVELD2 MIR96 MITF MPZ MSRB3 MYH14 MYH9 MYO15A MYO1A MYO1E MYO3A MYO6 MYO7A NDP NDRG1 NEFL NELL2 NF2 OPA1 OTOA OTOF OTOG OTOGL P2RX2 PABPN1 PAX3 PCDH15 PCDH9 PDZD7 PMP22 PNPT1 POLR1C POLR1D POU3F4 POU4F3 PROK2 PROKR2 PRPS1 PTPN11 PTPRQ PTPRR RDX RPGR SALL1 SALL4 SEC23A SEMA3E SERPINB6 SIX1 SIX5 SLC17A8 SLC19A2 SLC26A4 SLC26A5 SMAD4 SMPX SNAI2 SOX10 STRC TBC1D24 TCIRG1 TCOF1 TECTA TIMM8A TJP2 TMC1 TMEM126A TMIE TMPRSS3 TMPRSS4 TNC TPRN TRIOBP TRMU TSPEAR TYR USH1C USH1G USH2A WFS1 Table S2 Six mitochondrial deafness-related regions Locus Location (RefSeq: NC_012920.1) MT-RNR1 chrM:648-1601 MT-TL1 chrM:3230-3304 MT-CO1 chrM:5904-7445 MT-TS1 chrM:7446-7514 MT-TK chrM:8295-8364 MT-TE chrM:14674-14742 Table S3 Three deafness-related miRNAs Name Chromosome Position (hg19) miR-96 chr7: 129414532-129414609 miR-182 chr7: 129410223-129410332 miR-183 chr7: 129414745-129414854 Gao X, et al.