C Copyright 2017 Joshua James Goings
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Arxiv:1703.07468V1 [Cond-Mat.Str-El] 21 Mar 2017 Generating Spin Ice and Its Emergent Magnetic Monopoles Tion Metal fluoride Pyrochlores](https://docslib.b-cdn.net/cover/2089/arxiv-1703-07468v1-cond-mat-str-el-21-mar-2017-generating-spin-ice-and-its-emergent-magnetic-monopoles-tion-metal-uoride-pyrochlores-122089.webp)
Arxiv:1703.07468V1 [Cond-Mat.Str-El] 21 Mar 2017 Generating Spin Ice and Its Emergent Magnetic Monopoles Tion Metal fluoride Pyrochlores
Single-ion properties of the Seff = 1/2 XY antiferromagnetic pyrochlores, 0 0 2+ 2+ NaA Co2F7 (A = Ca , Sr ) K.A. Ross,1, 2 J.M. Brown,1 R.J. Cava,3 J.W. Krizan,3 S. E. Nagler,4 J.A. Rodriguez-Rivera,5, 6 and M. B. Stone4 1Colorado State University, Fort Collins, Colorado, 80523, USA 2Quantum Materials Program, Canadian Institute for Advanced Research, Toronto, Ontario M5G 1Z8, Canada∗ 3Princeton University, Princeton, New Jersey 08544, USA 4Quantum Condensed Matter Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA 5NIST Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA 6Materials Science and Engineering, University of Maryland, College Park, MD 20742, USA (Dated: March 23, 2017) The antiferromagnetic pyrochlore material NaCaCo2F7 is a thermal spin liquid over a broad 2+ temperature range (≈ 140 K down to TF = 2:4K), in which magnetic correlations between Co dipole moments explore a continuous manifold of antiferromagnetic XY states.1 The thermal spin liquid is interrupted by spin freezing at a temperature that is ∼ 2 % of the mean field interaction strength, leading to short range static XY clusters with distinctive relaxation dynamics. Here we report the low energy inelastic neutron scattering response from the related compound NaSrCo2F7, confirming that it hosts the same static and dynamic spin correlations as NaCaCo2F7. We then present the single-ion levels of Co2+ in these materials as measured by inelastic neutron scattering. An intermediate spin orbit coupling model applied to an ensemble of trigonally distorted octahedral crystal fields accounts for the observed transitions. -

Topological Insulators: Preliminaries1
PHYS598PTD A.J.Leggett 2013 Lecture 21 Topological insulators: preliminaries 1 Topological insulators: preliminaries1 At least as far as currently known, a good qualitative understanding of the properties of the class of materials now known as topological insulators (TI's) can be achieved on the basis of a picture of noninteracting electrons subject to a particular kind of band structure, which generally speaking involves nontrivial effects of the spin-orbit interaction (SOI). (As a result, the experimentally observed TI's generally contain heavy elements where the SOI is important). Thus, with the virtue of (considerable!) hindsight, the theory of TI's may be regarded as simply traditional undergraduate solid-state physics seen in a new light; the \new light" focuses particularly on states of the surface of the system, which have not traditionally received much attention. Let's start by reviewing some relevant elementary considerations. First, let's review some basic properties of a system which is confined to a 2-dimensional Hilbert space. Any such system is formally equivalent to a particle of spin 1=2 , and we will say that it is described by a \pseudospin" vector σ^, such that any pure state is an eigenstate of the operator n ·σ^ with eigenvalue ±1, where n is some unit vector: n · σ^j i = ±| i (1) We will usually choose to work in the basis of eigenstates ofσ ^z; depending on the context, these may be actual eigenstates of the true spin (intrinsic angular momentum) of the electron in question, but in the context of the theory of TI's are at least as as likely to correspond to different Bloch bands (cf. -

Spontaneous Rotational Symmetry Breaking in a Kramers Two- Level System
Spontaneous Rotational Symmetry Breaking in a Kramers Two- Level System Mário G. Silveirinha* (1) University of Lisbon–Instituto Superior Técnico and Instituto de Telecomunicações, Avenida Rovisco Pais, 1, 1049-001 Lisboa, Portugal, [email protected] Abstract Here, I develop a model for a two-level system that respects the time-reversal symmetry of the atom Hamiltonian and the Kramers theorem. The two-level system is formed by two Kramers pairs of excited and ground states. It is shown that due to the spin-orbit interaction it is in general impossible to find a basis of atomic states for which the crossed transition dipole moment vanishes. The parametric electric polarizability of the Kramers two-level system for a definite ground-state is generically nonreciprocal. I apply the developed formalism to study Casimir-Polder forces and torques when the two-level system is placed nearby either a reciprocal or a nonreciprocal substrate. In particular, I investigate the stable equilibrium orientation of the two-level system when both the atom and the reciprocal substrate have symmetry of revolution about some axis. Surprisingly, it is found that when chiral-type dipole transitions are dominant the stable ground state is not the one in which the symmetry axes of the atom and substrate are aligned. The reason is that the rotational symmetry may be spontaneously broken by the quantum vacuum fluctuations, so that the ground state has less symmetry than the system itself. * To whom correspondence should be addressed: E-mail: [email protected] -1- I. Introduction At the microscopic level, physical systems are generically ruled by time-reversal invariant Hamiltonians [1]. -

A Quantum Liquid of Magnetic Octupoles on the Pyrochlore Lattice
A quantum liquid of magnetic octupoles on the pyrochlore lattice Romain Sibille1,*, Nicolas Gauthier2, Elsa Lhotel3, Victor Porée1, Vladimir Pomjakushin1, Russell A. Ewings4, Toby G. Perring4, Jacques Ollivier5, Andrew Wildes5, Clemens Ritter5, Thomas C. Hansen5, David A. Keen4, Gøran J. Nilsen4, Lukas Keller1, Sylvain Petit6,* & Tom Fennell1 1Laboratory for Neutron Scattering and Imaging, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland. 2Stanford Institute for Materials and Energy Science, SLAC National Accelerator Laboratory and Stanford University, Menlo Park, California 94025, USA. 3Institut Néel, CNRS– Université Joseph Fourier, 38042 Grenoble, France. 4ISIS Pulsed Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK. 5Institut Laue-Langevin, 71 avenue des Martyrs, 38000 Grenoble, France. 6LLB, CEA, CNRS, Université Paris-Saclay, CEA Saclay, 91191 Gif-sur-Yvette, France. *email: [email protected] ; [email protected] Spin liquids are highly correlated yet disordered states formed by the entanglement of magnetic dipoles1. Theories typically define such states using gauge fields and deconfined quasiparticle excitations that emerge from a simple rule governing the local ground state of a frustrated magnet. For example, the ‘2-in-2-out’ ice rule for dipole moments on a tetrahedron can lead to a quantum spin ice in rare-earth pyrochlores – a state described by a lattice gauge theory of quantum electrodynamics2-4. However, f-electron ions often carry multipole degrees of freedom of higher rank than dipoles, leading to intriguing behaviours and ‘hidden’ orders5-6. Here we show that the correlated ground state of a Ce3+- based pyrochlore, Ce2Sn2O7, is a quantum liquid of magnetic octupoles. -

Magnetism in Uranium and Praseodymium Intermetallic Compounds Studied by Inelastic Neutron Scattering
Magnetism in uranium and praseodymium intermetallic compounds studied by inelastic neutron scattering 1=XJCL " =□ i— ' r~~ ~ 3 University College London A thesis submitted in accordance with the requirements of the University of London for the degree of Doctor of Philosophy Jonathan Paul Allen 2003 UMI Number: U602491 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. Dissertation Publishing UMI U602491 Published by ProQuest LLC 2014. Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Abstract The dispersion of magnetic excitations in PrNiSn has been studied using inelastic neutron scattering. The dispersion relations along [0 0 Q] and [Q 0 0] have been determined for the excitations observed at 1.5, 2.5 and 5.2 meV. The two lower excitations are proposed to be modes originating from the 2 meV level predicted by earlier inelastic measurements. Additional measurements of the 3.5 meV excitation already studied were performed to provide higher resolution data and improved statistics. Preliminary measurements on NdNiSn were also made. Searches have been made using inelastic neutron scattering to look for crystalline electric field (CEF) excitations in the uranium-based intermetallics UPdSn, UCu 2 Sn and U 3Pd 2 oSi 6 . -

Chapter 1 Magnetism of the Rare-Earth Ions in Crystals
Chapter 1 Magnetism of the Rare-Earth Ions in Crystals 3 1. Magnetism of the Rare-Earth ions in Crystals In this chapter we present the basic facts regarding the physics of magnetic phenomena of rare-earth ions in crystals. We discuss the theory of quantized angular momentum and the theory of the crystal field (CF) splitting of the electronic energy levels of the rare-earth ions in these crystals as required to give a better understanding for the material discussed in detail in later chapters in this book. Additional data and analyses can be found in corresponding references, which expand on topics that are discussed in each of the chapters in this book. 1.1. Electronic Structure and Energy Spectra of the “Free” Rare-Earth Ions In rare earth (RE) compounds, the lanthanide ions (from Ce to Yb) are usually found in the trivalent state RE3+. The ground electronic configuration of the RE ions may be written as [Xe]4f n, where [Xe] = 1s22s22p63s23p63d104s24p64d10 5s2p6 is the closed-shell configuration of the noble gas xenon, and n is the number of electrons in the unfilled 4f n shell, ranging from n = 1 for Ce3+ to n = 13 for Yb3+. The characteristic magnetic moment for each RE ion leads to an interac- tion between that ion and an applied external magnetic field H, producing interesting magnetic and magnetooptical fea- tures in the RE compounds. Over the years, methods of numerical analysis have been developed to calculate the energy states of “free” RE ions (that is, for ions that are not in a ligand or crystal-field environment). -

The Pin Groups in Physics: C, P, and T
IHES-P/00/42 The Pin Groups in Physics: C, P , and T Marcus Berga , C´ecile DeWitt-Morettea, Shangjr Gwob and Eric Kramerc aDepartment of Physics and Center for Relativity, University of Texas, Austin, TX 78712, USA bDepartment of Physics, National Tsing Hua University, Hsinchu 30034, Taiwan c290 Shelli Lane, Roswell, GA 30075 USA arXiv:math-ph/0012006v1 6 Dec 2000 Abstract A simple, but not widely known, mathematical fact concerning the coverings of the full Lorentz group sheds light on parity and time reversal transformations of fermions. Whereas there is, up to an isomorphism, only one Spin group which double covers the orientation preserving Lorentz group, there are two essentially different groups, called Pin groups, which cover the full Lorentz group. Pin(1,3) is to O(1,3) what Spin(1,3) is to SO(1,3). The existence of two Pin groups offers a classification of fermions based on their properties under space or time reversal finer than the classification based on their properties under orientation preserving Lorentz transformations — provided one can design experiments that distinguish the two types of fermions. Many promising experimental setups give, for one reason or another, identical results for both types of fermions. These negative results are reported here because they are instructive. Two notable positive results show that the existence of two Pin groups is relevant to physics: In a neutrinoless double beta decay, the neutrino emitted and reabsorbed in the course • of the interaction can only be described in terms of Pin(3,1). If a space is topologically nontrivial, the vacuum expectation values of Fermi currents • defined on this space can be totally different when described in terms of Pin(1,3) and Pin(3,1). -

Time Reversal I Operator
Time reversal operator Masatsugu Sei Suzuki, Department of Physics State University of New York at Binghamton (Date: December 08, 2016) If a movie film is run backwards, all movement reversed can be seen: a falling stone will be replaced by a stone projected upwards. This is true because Newton’s equation of motion in a uniform gravitational field is unchanged when the time t is replaced by –t. However, in the Schrödinger picture of quantum mechanics, the simple time inversion operator t → −t also changes the sign of the energy, because the energy operator is given by ih ∂ /( ∂t) . Thus in quantum mechanics, the time inversion operation has a much more drastic effect than simply reversing the motion. Since the idea of reversing the motion is perfectly sensible in classical mechanics, i.e., one which reverses directions of motion but does not change the sign of the energy. Here we construct such an operation, called time-reversal and denoted by Θˆ . It will turn out that Θˆ is a very unusual operator, being non-linear. Θˆ = UKˆˆ (Uˆ and Kˆ decomposition) where Θˆ is an anti-unitary operator, Uˆ is the unitary operator, and Kˆ is the complex conjugate operator. _____________________________________________________________________________ H.A. Kramers Hendrik Anthony "Hans" Kramers (2 February 1894 – 24 April 1952) was a Dutch physicist who worked with Niels Bohr to understand how electromagnetic waves interact with matter. 1 https://en.wikipedia.org/wiki/Hans_Kramers _____________________________________________________________________________ 1. Classical mechanics The time-reversal invariance is the only one that can be discussed within the framework of classical theory. -
Quantum Tunnelling of the Magnetisation in Molecular Nanomagnets
FEATURES Quantum tunnelling of the magnetisation in molecular nanomagnets R. Sessoli Department ofChemistry, University ofFlorence and INSTM, 50019 Sesto Fiorentino, Italy he twmel effect represents the fascinating point where quan in fact due to the strong dependence of tunnelling on the axial T tum mechanics meets the classical world. Even though not and transverse magnetic anisotropy that, on their turn, are relat very easily observable, quantum tunnelling in charge transport is ed to the dimensions of the nanoparticle. Ensembles of a well known phenomenon, now even exploited in devices, as in nanoparticles are unfortunatelycharacterisedbya distribution in the Super Conducting Quantum Interference Device (SQUID). size and shape. On the contrary the twmelling phenomenon in magnetic mate rials is much less investigated. Most ofthe properties that make Molecular nanomagnets magnetic materials so important in our every-day life, i. e. mag In the beginning ofthe nineties chemists, and in particular mole netic hysteresis and related memory effect, do not reveal any cular chemists, entered into the game bystartingto synthesise and macroscopic quantum effect. However, ifthe dimensions ofthe investigate objects that can be seen as the"missing link" between magnetic material are shrunkbelow the width ofa domain wall, the quantum word ofparamagnetic metal centres and the classi all the magnetic moments move coherently and the process of cal one ofmagnetic particles. These new materials, known as magnetisation reversal occurs by the overcoming of an energy molecular nanomagnets or as Single Molecule Magnets, are barrier originated by the magnetic anisotropy ofthe material. indeed clusters comprising a relatively small number ofpara This barriercanbecrossed through a thermally activated process, magnetic centres. -

Studies of Oriented Aromatic Hydrocarbons in the Phos Phorescent State at Low Magnetic Fields
WINER, Arthur Melvyn, 1942- ELECTRON PARAMAGNETIC RESONANCE STUDIES OF ORIENTED AROMATIC HYDROCARBONS IN THE PHOS PHORESCENT STATE AT LOW MAGNETIC FIELDS. The Ohio State University, Ph.D., 1969 Chemistry, physical University Microfilms, Inc., Ann Arbor, Michigan THIS DISSERTATION HAS BEEN MICROFILMED EXACTLY AS RECEIVED ELECTRON PARAMAGNETIC RESONANCE STUDIES OF ORIENTED AROMATIC HYDROCARBONS IN THE PHOSPHORESCENT STATE AT LOW MAGNETIC FIELDS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Arthur Melvyn Winer, B.S, ***** The Ohio State University 1969 Approved by Adviser Department of Chemistry To My Father ii ACKNOWLEDGMENTS It is a pleasure to acknowledge the contribution to this work of Professor Roger E. Gerkin, who not only pro vided direction and encouragement throughout the research, for which I am grateful, but who also established an atmos phere of conviviality which made my tenure as his student most enjoyable. Valuable discussions with Mr. William J. Rogers, Mr. Peter Szerenyi and Mr. David L. Thorsell of this labora tory and with Professor Clyde A. Hutchison of the Univer sity of Chicago are gratefully acknowledged. I wish to thank Mr. Thorsell also for taking the photographs of research equipment which appear in the dissertation. The work of Mr. Terence Ireland in maintaining and improving the electronic equipment was essential to the completion of this research and was greatly appreciated. I wish to thank Professor Jack G. Calvert, Dr. Susan Collier and Dr. T. Navaneeth Rao for use of the Turner spectrophotometer, and Professor Peter W. R. -
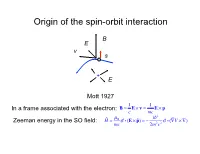
Origin of the Spin-Orbit Interaction
Origin of the spin-orbit interaction B E v s + E Mott 1927 1 1 In a frame associated with the electron: B = E × v = E × p c mc 2 ˆ µB i Zeeman energy in the SO field: H = σ i (E × pˆ ) = − 2 2 σ i (∇V × ∇) mc 2m c 1/c expansion of the Dirac equation pˆ 2 pˆ 4 2 ˆ 2 ˆ H = + eV + 2 2 + 2 2 ∇ V + 2 2 σi(∇V × p) 2m 8mc 8mc 4mc non-relativistic K.E. correction Darwin term SOI All three corection terms are of the same order Smaller by a factor of 2 (“Thomson factor of two”, 1926) Reason: non-inertial frame Landau& Lifshits IV: Berestetskii, Lifshits, Pitaevskii Quantum Electrodynamics, Ch. 33 2 SO coupling (Ze2 / c) 2 2 (Ze / c) Si14 Ga31 Ge32 As33 Bi83 0.01 0.051 0.055 0.058 0.38 Ga32 Ga33 Si14 Ge32 Bi83 Fine structure of atomic levels Hydrogen 2s Runge-Lentz (non-relativistic Coulomb) 2s1/2 +spin degeneracy Johnson-Lippman (relativistic Coulomb) degeneracy lifted by radiation corrections (Lamb shift) Two types of SOI in solids 1) Symmetry-independent: exists in all types of crystals stem from SOI in atomic orbitals 2) Symmetry-dependent: exists only in crystals without inversion symmetry a) Dresselhaus interaction (bulk): Bulk-Induced-Assymetry (BIA) b) Bychkov-Rashba (surface): Surface-Induced-Asymmetry (SIA) Example of symmetry-independent SOI: SO-split-off valence bands Winkler, Ch. 3 I E: j = 1 / 2; jz = ±1 / 2 no I 2 2 2 ⎡(ZGa + ZAs ) / 2 ⎤ ⎛ ZGe ⎞ ⎛ 32⎞ ⎢ ⎥ = ⎜ ⎟ = ⎜ ⎟ = 5.2 ⎣ ZSi ⎦ ⎝ ZSi ⎠ ⎝ 14 ⎠ ΔGaAs / ΔSi = .34/0.044 ≈ 7.7 ΔGaAs / ΔGe = .34/.029 ≈ 1.2 Non-centrosymmetric crystals 7 space group: Oh (non-symmorphic) factor group Oh = Td × I S. -

General Physics Questions
GENERAL PHYSICS QUESTIONS 1. Newton’s laws of motion, limits of their applicability, relativistic dynamics. 2. Law of universal gravitation, Kepler’s laws. 3. Conservation laws in physics. 4. Mechanical properties of material media: liquids, elastic media, mechanical waves. 5. Laws of thermodynamics. 6. Phase transitions, classification of phase transitions, examples. 7. Statistical description of many-body systems, statistical ensembles. 8. Electromagnetic field, Maxwell’s equations of the electromagnetic field. 9. Invariance of electrodynamics under Lorentz transformations, fundamentals of the special theory of relativity. 10. Electric and magnetic fields in material media, dielectric polarization, conductivity, magnetization. 11. Electromagnetic waves, dispersion, polarization, reflection and refraction of waves, diffraction and interference of waves, coherence. 12. Doppler effect for acoustic and electromagnetic waves. 13. Experimental basis of quantum mechanics, the uncertainty principle and its consequences. 14. Schrödinger equation, quantum harmonic oscillator, single-electron atom. 15. Approximate methods in quantum mechanics: perturbation theory, variational approach. 16. Periodic table of elements, chemical bonds, basic properties of molecules. 17. Electronic structure of solids, semiconductors, conductors, insulators. 18. Physical underpinnings of optical spectroscopy; atomic spectra, molecular spectra. 19. Black-body radiation. 20. Indistinguishability of elementary particles; spin-statistics relation. Ideal gas of bosons or fermions. 21. Structure of atomic nuclei and nuclear reactions; binding energy of a nucleus, radioactive decay. 22. Classification of elementary particles; quarks and leptons, structure of hadrons. PROFILE-SPECIFIC QUESTIONS Solid state physics profile Block A: Semiconductors, metals and x-ray spectroscopy in solid state physics 1. Crystalline structure and chemical bonds in solids. 2. Band structure of solids – the tight binding model and the quasi-free electron model; Bloch theorem; electronic density of states.