In Re Delcath Systems, Inc. Securities Litigation 13-CV-03116-Amended
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Drug Delivery Technology Y
* DDT Nov-Dec 2007 Working 11/9/07 2:29 PM Page 1 November/December 2007 Vol 7 No 10 IN THIS ISSUE Company Profiles 12 Drug Delivery Technologies 58 Excipients, Polymers, Liposomes & Lipids 78 Contract Pharmaceutical & Biological Development Services 83 Machinery & Laboratory Equipment and Software 96 Technology Showcase 102 The science & business of specialty pharma, biotechnology, and drug delivery www.drugdeliverytech.com * DDT Nov-Dec 2007 Working 11/9/07 2:39 PM Page 2 * DDT Nov-Dec 2007 Working 11/9/07 2:40 PM Page 3 * DDT Nov-Dec 2007 Working 11/9/07 2:40 PM Page 4 November/December 2007 Vol 7 No 10 PUBLISHER/PRESIDENT Ralph Vitaro EXECUTIVE EDITORIAL DIRECTOR Dan Marino, MSc [email protected] CREATIVE DIRECTOR Shalamar Q. Eagel CONTROLLER Debbie Carrillo CONTRIBUTING EDITORS Cindy H. Dubin Debra Bingham Jason McKinnie TECHNICAL OPERATIONS Mark Newland EDITORIAL SUPPORT Nicholas D. Vitaro ADMINISTRATIVE SUPPORT Kathleen Kenny Corporate/Editorial Office 219 Changebridge Road, Montville, NJ 07045 Tel: (973)299-1200 Fax: (973) 299-7937 www.drugdeliverytech.com Advertising Sales Offices East & Midwest Victoria Geis - Account Executive Coming in 2008 Cheryl S. Stratos - Account Executive 103 Oronoco Street, Suite 200 Alexandria, VA 22314 Tel: (703) 212-7735 Drug Delivery Weekly & Fax: (703) 548-3733 E-mail: [email protected] Specialty Pharma News E-mail: [email protected] West Coast Warren De Graff Western Regional Manager 818 5th Avenue, Suite 301 San Rafael, CA 94901 Tel: (415) 721-0644 Fax: (415) 721-0665 E-mail: [email protected] 0 The weekly electronic newsletter from the publishers of Drug 1 International o N Delivery Technology and Specialty Pharma will provide over 12,000 Ralph Vitaro 7 219 Changebridge Road l o subscribers with the latest news of business deals, alliances, and V Montville, NJ 07045 Tel: (973) 299-1200 7 technology breakthroughs from the pharmaceutical, specialty 0 Fax: (973) 299-7937 0 2 pharmaceutical, drug delivery, and biotechnology industries. -

Chemosaturation with Percutaneous Hepatic Perfusion in Unresectable Hepatic Metastases Evan S
Special Report Chemosaturation With Percutaneous Hepatic Perfusion in Unresectable Hepatic Metastases Evan S. Glazer, MD, PhD, and Jonathan S. Zager, MD Background: In patients with hepatic metastases from solid-organ malignancies, surgical resection may be a potentially curative option, but it is not possible in most cases. Chemosaturation with percutaneous hepatic perfusion was developed for the management of unresectable metastases to the liver. Methods: Relevant medical literature was summarized with regard to the outcomes and limitations of che- mosaturation with percutaneous hepatic perfusion. Results: Six articles were identified that contained data on 91 individuals who received chemosaturation with percutaneous hepatic perfusion. More than 60% of these study patients were diagnosed with ocular melanoma. The overall response rate was 48% and the rate of disease control was 90%. Chemosaturation with percutaneous hepatic perfusion improved the rates of overall and hepatic progression-free survival (PFS). The data are limited but suggest that the rate of PFS was improved in study patients with isolated melanoma hepatic metastases who received chemosaturation with percutaneous hepatic perfusion compared with those assigned to standard care. Conclusions: Our results suggest that chemosaturation with percutaneous hepatic perfusion produces favor- able tumor response rates in select individuals with unresectable hepatic metastases from multiple primary cancers, particularly ocular and cutaneous melanomas. Data from a single randomized -

Delcath Systems Schedules Conference Call to Report 2021 Second Quarter Financial Results
Source: Delcath Systems, Inc. July 28, 2021 09:00 ET Delcath Systems Schedules Conference Call to Report 2021 Second Quarter Financial Results NEW YORK, July 28, 2021 (GLOBE NEWSWIRE) -- Delcath Systems, Inc. (Nasdaq: DCTH ), an interventional oncology company focused on the treatment of rare primary and metastatic cancers of the liver, announced today it will host a conference call on August 10, 2021 at 8:30 AM Eastern Time to discuss results for its second quarter ended June 30, 2021. Conference Call Information To participate in this event, dial approximately 5 to 10 minutes before the beginning of the call. Date: August 10, 2021 Time: 8:30 AM Eastern Time Toll Free: 877-545-0523; Entry Code: 148562 International: 973-528-0016; Entry Code: 148562 The call will also be available over the Internet and accessible at: https://www.webcaster4.com/Webcast/Page/2475/42376 About Delcath Systems, Inc. Delcath Systems, Inc. is an interventional oncology company focused on the treatment of primary and metastatic liver cancers. The company’s proprietary percutaneous hepatic perfusion (PHP) system is designed to administer high-dose chemotherapy to the liver while controlling systemic exposure and associated side effects. In the United States, the PHP system is being developed under the tradename HEPZATO KIT (melphalan hydrochloride for injection/hepatic delivery system), or HEPZATO, and is considered a combination drug and device product regulated by the United States Food and Drug Administration (FDA). In Europe, the PHP system is regulated as a Class IIb medical device and is approved for sale under the trade name CHEMOSAT Hepatic Delivery System for Melphalan, or CHEMOSAT, where it has been used at major medical centers to treat a wide range of cancers of the liver. -

Delcath Systems, Inc
Delcath Systems, Inc. Corporate Presentation (NASDAQ: DCTH) August 2020 Forward-looking Statements This presentation contains forward-looking statements, which are subject to certain risksand uncertainties that can cause actual results to differ materially from those described. Factors that may cause such differences include, but are not limited to, uncertainties relating to: the timing and results of the Company’s clinical trials including without limitation the OM and ICC clinical trial programs, timely enrollment and treatment of patients in the global Phase 3 OM and ICC Registration trials, IRB or ethics committee clearance of the Phase 3 OM and ICC Registration trial protocols from participating sites andthe timing of site activation and subject enrollment in each trial, the impact of the presentations at major medical conferences and future clinical results consistent with the data presented, the Company’s ability to successfully commercialize the Melphalan HDS/CHEMOSAT system and the potential of the Melphalan HDS/CHEMOSAT system as a treatment for patients with primary and metastatic disease in the liver, our ability to obtain reimbursement for the CHEMOSAT system in various markets, approval of the current or future Melphalan HDS/CHEMOSAT system for delivery and filtration of melphalan or other chemotherapeutic agents for various indications in the U.S. and/or in foreign markets, actions by the FDA or other foreign regulatory agencies, the impact of the Company’s exclusive licensing agreement with medac on commercial adoption in Europe and resulting revenue, if any, the Company’s ability to successfully enter into other strategic partnerships and distribution arrangements in foreign markets and the timing and revenue, if any, of the same, uncertainties relating to the timing and results of research and development projects, and uncertainties regarding the Company’s ability to obtain financial and other resources for any research, development, clinical trials and commercialization activities. -

Evaluation of Melphalan, Oxaliplatin, and Paclitaxel in Colon, Liver, And
ANTICANCER RESEARCH 33: 1989-2000 (2013) Evaluation of Melphalan, Oxaliplatin, and Paclitaxel in Colon, Liver, and Gastric Cancer Cell Lines in a Short-term Exposure Model of Chemosaturation Therapy by Percutaneous Hepatic Perfusion RAJNEESH P. UZGARE, TIMOTHY P. SHEETS and DANIEL S. JOHNSTON Pharmaceutical Research and Development, Delcath Systems, Inc., Queensbury, NY, U.S.A. Abstract. Background: The goal of this study was to candidates for use in the CS-PHP system to treat patients determine whether liver, gastric, or colonic cancer may be with gastric and colonic metastases, and primary cancer of suitable targets for chemosaturation therapy with the liver. percutaneous hepatic perfusion (CS-PHP) and to assess the feasibility of utilizing other cytotoxic agents besides Chemotherapeutic molecules exert beneficial clinical effects melphalan in the CS-PHP system. Materials and Methods: by inhibiting cell growth or by inducing cell death via Forty human cell lines were screened against three cytotoxic apoptosis. They can be divided into several categories based chemotherapeutic agents. Specifically, the dose-dependent on their mechanisms of action. The chemotherapeutic agent effect of melphalan, oxaliplatin, and paclitaxel on melphalan hydrochloride, which has been approved by the proliferation and apoptosis in each cell line was evaluated. US Food and Drug Administration and is used in the These agents were also evaluated for their ability to induce treatment of multiple myeloma and ovarian cancer, is a apoptosis in normal primary human hepatocytes. A high- derivative of nitrogen mustard that acts as a bifunctional dose short-term drug exposure protocol was employed to alkylating agent. Melphalan causes the alkylation of DNA at simulate conditions encountered during CS-PHP. -

Core Messages
1 DELCATH SYSTEMS, INC CHEMOSAT® 2 DELCATH SYSTEMS, INC CHEMOSAT® 3 DELCATH SYSTEMS, INC CHEMOSAT® University of Maryland-Baltimore 4 DELCATH SYSTEMS, INC CHEMOSAT® Unresectable cancers (primary or metastatic) confined to liver are a significant clinical problem: Colorectal cancer: 35,000/yr HCC/cholangiocarcinoma: 20,500/yr Ocular melanoma: 2,500/yr Neuroendocrine tumors: 3,000/yr Other histologies Therapeutic options are limited and survival after diagnosis of diffuse liver metastases is short. Morbidity and mortality in this setting is most frequently a consequence of disease progression in the liver. Effective control of isolated diffuse metastases to the liver is a significant clinical challenge 5 DELCATH SYSTEMS, INC CHEMOSAT® Intra-arterial delivery of therapeutic agents (versus portal venous system) will be preferentially delivered to metastases Dose escalation of therapeutic agents is possible Normal tissue tolerance of the perfused organ is higher Infused agent should have a broad non-specific mechanism of action Melphalan has a broad spectrum of activity with dose levels achieved during PHP Control of systemic exposure is possible First pass effect (FUDR) Sequestration in the liver (TACE, SIRS) Physical isolation Filtration Systemic toxicities can be easily diagnosed and managed; comparable to the toxicities seen with commonly used systemic chemotherapeutic agents Potentially provides treatment to the entire organ Provides opportunity to use novel agents 6 DELCATH SYSTEMS, INC CHEMOSAT® Old Linear Model -

Percutaneous Hepatic Perfusion with Melphalan in Patients with Unresectable Ocular Melanoma Metastases Confined to the Liver: a Prospective Phase II Study
Ann Surg Oncol https://doi.org/10.1245/s10434-020-08741-x ORIGINAL ARTICLE – PERITONEAL SURFACE MALIGNANCY Percutaneous Hepatic Perfusion with Melphalan in Patients with Unresectable Ocular Melanoma Metastases Confined to the Liver: A Prospective Phase II Study T. Susanna Meijer, MD1 , Mark C. Burgmans, MD, PhD1, Eleonora M. de Leede, MD2, Lioe-Fee de Geus-Oei, MD, PhD1,3, Bas Boekestijn, MD1, Henricus J. M. Handgraaf, MD, PhD2, Denise E. Hilling, MD, PhD2, Jacob Lutjeboer1, Jaap Vuijk, MD, PhD4, Christian H. Martini, MD, PhD4, Arian R. van Erkel, MD, PhD1, Rutger W. van der Meer, MD, PhD1, Fred G. J. Tijl5, Frank M. Speetjens, MD, PhD6, Ellen Kapiteijn, MD, PhD6, and Alexander L. Vahrmeijer, MD, PhD2 1Department of Radiology, Leiden University Medical Center, Leiden, The Netherlands; 2Department of Surgery, Leiden University Medical Center, Leiden, The Netherlands; 3Biomedical Photonic Imaging Group, University of Twente, Enschede, The Netherlands; 4Department of Anesthesiology, Leiden University Medical Center, Leiden, The Netherlands; 5Department of Extra Corporal Circulation, Leiden University Medical Center, Leiden, The Netherlands; 6Department of Medical Oncology, Leiden University Medical Center, Leiden, The Netherlands ABSTRACT (BOR). Secondary endpoints included overall survival Background. Ocular melanoma is the most common pri- (OS), progression-free survival (PFS), hepatic PFS (hPFS), mary intraocular malignancy and has a very poor prognosis and safety. once liver metastases occur. Theaim of this study was to Results. Sixty-four M-PHP procedures were performed in prospectively assess the efficacy and safety of percutaneous 35 patients between February 2014 and June 2017. The hepatic perfusion with melphalan (M-PHP) using the new ORR was 72%. -

View Annual Report
UNITED STATES SECURITIES AND EXCHANGE COMMISSION WASHINGTON, D.C. 20549 FORM 10-K x Annual report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 for the fiscal year ended December 31, 2009 o Transition report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 for the transition period from ____________ to ____________ Commission file number: 001-16133 DELCATH SYSTEMS, INC. Delaware 06-1245881 (State or other jurisdiction of incorporation or organization) (I.R.S. Employer Identification No.) 600 Fifth Avenue, 23rd Floor, New York, NY 10020 (Address of principal executive offices) (Zip Code) 212-489-2100 (Registrant’s telephone number, including area code) Securities registered pursuant to Section 12(b) of the Act: Title of Each Class Name of Each Exchange on Which Registered Common Stock, par value $0.01 per share The NASDAQ Stock Market LLC Securities registered pursuant to Section 12(g) of the Act: None. Indicate by check mark if the registrant is a well-known seasoned issuer, as defined by Rule 405 of the Securities Act. Yes o No x Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act. Yes o No x Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. -

Delcath Systems Announces Oral Presentation at the American Society of Clinical Oncology (ASCO) Annual Meeting
Delcath Systems Announces Oral Presentation at the American Society of Clinical Oncology (ASCO) Annual Meeting April 30, 2021 A Presentation of Positive Preliminary Efficacy Results from the FOCUS Phase III trial Company Q&A Webinar Scheduled for Monday, June 7, 2021 at 8:30am ET NEW YORK, April 30, 2021 (GLOBE NEWSWIRE) -- Delcath Systems, Inc. (Nasdaq: DCTH ), an interventional oncology company focused on the treatment of rare primary and metastatic cancers of the liver, today announced an oral presentation at the American Society of Clinical Oncology (ASCO) Annual Meeting to be held virtually June 4-8, 2021, and announced a company Q&A webinar has been scheduled for Monday, June 7, 2021 at 8:30am ET. ASCO Presentation Details: Title: Percutaneous hepatic perfusion (PHP) with melphalan for patients with ocular melanoma liver metastases: Preliminary results of FOCUS (PHP-OCM-301/301A) phase III trial. Type: Oral Abstract Presentation Track: Melanoma/Skin Cancers Presenter: Jonathan S. Zager, MD FACS Abstract Number: 9510 Date and Time: Available Starting June 4, 2021, 9:00 AM (EDT) Dr. Zager will be presenting on behalf of the Principal Investigators that participated in the FOCUS trial. The full ASCO meeting program is available at: www.asco.org. Q&A Webinar Details: The company invites investors to join a Q&A webinar on June 7 at 8:30am ET, hosted by Gerard Michel, CEO. Mr. Michel will introduce the company and its strategy, as well as discuss the positive preliminary results from its Phase 3 FOCUS Trial of HEPZATO in patients with metastatic ocular melanoma. Management will also provide answers on the live call to any questions submitted by attendees. -

Delcath Systems to Present at Access to Giving Virtual Conference on July 13, 2021 at 3 P.M
Delcath Systems to Present at Access to Giving Virtual Conference on July 13, 2021 at 3 p.m. ET July 7, 2021 NEW YORK, July 07, 2021 (GLOBE NEWSWIRE) -- Delcath Systems, Inc. (Nasdaq: DCTH), an interventional oncology company focused on the treatment of rare primary and metastatic cancers of the liver, announced today that the company will present at Access to Giving Virtual Conference. The presentation will be given by Delcath’s Chief Executive Officer, Gerard Michel. The conference is free to all registrants. Registration Details Date: Tuesday, July 13, 2021 Time: 3 p.m. ET Link: https://access-to-giving.events.issuerdirect.com/signup To learn more about the event or to schedule a one-on-one meeting with Delcath’s management, please visit https://www.accesstogiving.com/ or email [email protected]. About Access to Giving Virtual Conference Access to Giving is a first-of-its-kind virtual investor conference where companies will have the opportunity to present their story and conduct 1x1 meetings with qualified investors, for charity. Investors will make donations to purchase a block of meetings to meet with companies. About Delcath Systems, Inc. Delcath Systems, Inc. is an interventional oncology company focused on the treatment of primary and metastatic liver cancers. Our investigational product, HEPZATO KIT (melphalan hydrochloride for injection/hepatic delivery system), is designed to administer high-dose chemotherapy to the liver while controlling systemic exposure and associated side effects. HEPZATO KIT has not been approved by the U.S. Food & Drug Administration (FDA) for sale in the U.S. In Europe, our system is marketed under the trade name Delcath CHEMOSAT® Hepatic Delivery System for Melphalan (CHEMOSAT) and has been CE Marked and used at major medical centers to treat a wide range of cancers of the liver. -
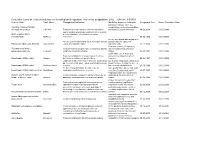
Designated Indication Cumulative List of All Products That Have Received
Cumulative List of all Products that have received Orphan Designation: Total active designations: 2002 Effecive: 5/5/2009 Generic Name Trade Name Designated Indication treatmentMarketing and/or Approved modification Indication of the Designated Date Market Exculsivity Date following conditions, which are Vaccinia Immune Globulin complications resulting from smallpox (Human) Intravenous CNJ-016 ForTreatment the control of complications and prevention of vacciniaof hemorrhagic vaccination episodes vaccination: Eczema vaccinatum 18.06.2004 01.05.9999 and for surgical prophylaxis in patients with hemophilia Antihemophilic factor A (congenital factor VIII deficiency or classic (recombinant) ReFacto hemophilia). 08.02.1996 01.01.9999 For use as a thyroid blocking agent in For use as a thyroid blocking agent in pediatric patients pediatric patients exposed to Potassium Iodide Oral Solution ThyroShield exposed to radiactive iodine radiactive iodine 17.11.2004 01.01.9999 Treatment (rescue) of respiratory Pulmonary surfactant For the treatment and prevention of respiratory distress distress syndrome in premature replacement, porcine Curosurf syndrome in premature infants. Longinfants. term treatment of children with 02.08.1993 01.01.9999 growth failure due to inadequate Treatment of idiopathic or organic growth hormone secretion of endogenous growth Somatropin (rDNA origin) Saizen deficiency in children with growth failure. Long-termhormone. treatment of growth failure 06.03.1987 01.01.9999 Long-term treatment of children who have growth failure due to a lack of adequate endogenous due to a lack of adequate endogenous growth hormone growth hormone secetion for once- or Somatropin (rDNA origin) Nutropin Depot secretion. twice-a-month administration. 28.10.1999 01.01.9999 Treatment of growth failure in children due to have growth failure due to inadequate Somatropin (rDNA origin) injection Norditropin inadequate growth hormone secretion. -

Percutaneous Hepatic Perfusion with CHEMOSAT Delivery System
cells Review A New Option for the Treatment of Intrahepatic Cholangiocarcinoma: Percutaneous Hepatic Perfusion with CHEMOSAT Delivery System Pier Francesco Ferrucci 1,* , Emilia Cocorocchio 2 , Guido Bonomo 3, Gianluca Maria Varano 3, Paolo Della Vigna 3 and Franco Orsi 3 1 Tumor Biotherapy Unit, Department of Experimental Oncology, European Institute of Oncology, IRCCS, 20141 Milan, Italy 2 Hematoncology Division, European Institute of Oncology, IRCCS, 20141 Milan, Italy; [email protected] 3 Interventional Radiology Division, European Institute of Oncology, IRCCS, 20141 Milan, Italy; [email protected] (G.B.); [email protected] (G.M.V.); [email protected] (P.D.V.); [email protected] (F.O.) * Correspondence: [email protected]; Tel.: +39-029-4371-094 Abstract: Liver metastases are a major management problem; since they occur in tumors of different origin, they are often multiple, difficult to visualize and can lie dormant for many years. Patients with liver metastases usually die of their disease, mostly due to liver failure, since systemic treatments are unable to eradicate micro-metastasis, and interventional loco-regional procedures cannot treat all existing ones. Cholangiocarcinoma (CCA) is the second most common primary liver tumor, showing a poor overall prognosis. When resection is not possible, treatment options include tumor-focused or local ablative therapy, organ-focused or regional therapy and systemic therapy. We reviewed available ® loco-regional therapeutic options, with particular focus on the CHEMOSAT Melphalan/Hepatic Delivery System (CS-HDS), which is uniquely positioned to perform a percutaneous hepatic perfusion Citation: Ferrucci, P.F.; Cocorocchio, (PHP), in order to treat the entire liver as a standalone or as complementary therapy.