A Myosin-7B Dependent Endocytosis Pathway Mediates Cellular Entry of Α-Synuclein Fibrils
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Myosin Myth4-FERM Structures Highlight Important Principles of Convergent Evolution
Myosin MyTH4-FERM structures highlight important principles of convergent evolution Vicente José Planelles-Herreroa,b, Florian Blanca,c, Serena Sirigua, Helena Sirkiaa, Jeffrey Clausea, Yannick Souriguesa, Daniel O. Johnsrudd, Beatrice Amiguesa, Marco Cecchinic, Susan P. Gilberte, Anne Houdussea,1,2, and Margaret A. Titusd,1,2 aStructural Motility, Institut Curie, CNRS, UMR 144, PSL Research University, F-75005 Paris, France; bUPMC Université de Paris 6, Institut de Formation Doctorale, Sorbonne Universités, 75252 Paris Cedex 05, France; cLaboratoire d’Ingénierie des Fonctions Moléculaires, Institut de Science et d’Ingénierie Supramoléculaires, UMR 7006 CNRS, Université de Strasbourg, F-67083 Strasbourg Cedex, France; dDepartment of Genetics, Cell Biology and Development, University of Minnesota, Minneapolis, MN 55455; and eDepartment of Biological Sciences, Rensselaer Polytechnic Institute, Troy, NY 12180 Edited by James A. Spudich, Stanford University School of Medicine, Stanford, CA, and approved March 31, 2016 (received for review January 15, 2016) Myosins containing MyTH4-FERM (myosin tail homology 4-band (Fig. 1). These MF myosins are widespread and likely quite an- 4.1, ezrin, radixin, moesin, or MF) domains in their tails are found cient because they are found in many different branches of the in a wide range of phylogenetically divergent organisms, such as phylogenetic tree (5, 6), including Opisthokonts (which includes humans and the social amoeba Dictyostelium (Dd). Interestingly, Metazoa, unicellular Holozoa, and Fungi), Amoebozoa, and the evolutionarily distant MF myosins have similar roles in the exten- SAR (Stramenopiles, Alveolates, and Rhizaria) (Fig. 1 A and B). sion of actin-filled membrane protrusions such as filopodia and Over the course of hundreds of millions years of parallel evolution bind to microtubules (MT), suggesting that the core functions of the MF myosins have acquired or maintained roles in the formation these MF myosins have been highly conserved over evolution. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Spectrum of MYO7A Mutations in an Indigenous South African
G C A T T A C G G C A T genes Article Spectrum of MYO7A Mutations in an Indigenous South African Population Further Elucidates the Nonsyndromic Autosomal Recessive Phenotype of DFNB2 to Include Both Homozygous and Compound Heterozygous Mutations Rosemary Ida Kabahuma 1,2,*, Wolf-Dieter Schubert 2, Christiaan Labuschagne 3, Denise Yan 4, Susan Halloran Blanton 4,5 , Michael Sean Pepper 6 and Xue Zhong Liu 4,5,* 1 Department of Otorhinolaryngology, University of Pretoria, Pretoria 0001, South Africa 2 Departments of Biochemistry, Genetics and Microbiology, Faculty of Natural and Agricultural Sciences, University of Pretoria, Pretoria 0001, South Africa; [email protected] 3 Inqaba Biotechnical Industries, Pretoria 0002, South Africa; [email protected] 4 Department Otolaryngology, University of Miami Miller School of Medicine, Miami, FL 33136, USA; [email protected] (D.Y.); [email protected] (S.H.B.) 5 Dr. John T. Macdonald Foundation Department of Human Genetics, and John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, Miami, FL 33136, USA 6 Department Immunology and SAMRC Extramural Unit for Stem Cell Research and Therapy, Faculty of Health Sciences, Institute for Cellular and Molecular Medicine, University of Pretoria, Citation: Kabahuma, R.I.; Pretoria 0001, South Africa; [email protected] Schubert, W.-D.; Labuschagne, C.; * Correspondence: [email protected] (R.I.K.); [email protected] (X.Z.L.) Yan, D.; Blanton, S.H.; Pepper, M.S.; Liu, X.Z. Spectrum of MYO7A Abstract: MYO7A gene encodes unconventional myosin VIIA, which, when mutated, causes a phe- Mutations in an Indigenous South notypic spectrum ranging from recessive hearing loss DFNB2 to deaf-blindness, Usher Type 1B African Population Further (USH1B). -

MYO7A Gene Myosin VIIA
MYO7A gene myosin VIIA Normal Function The MYO7A gene provides instructions for making a protein called myosin VIIA, which is part of a group of proteins called unconventional myosins. These proteins, which have similar structures, help transport molecules within cells. Myosins interact with actin, a protein that is important for cell movement and shape. Researchers believe that myosins use long filaments of actin as tracks along which to transport other molecules. Myosin VIIA is made in the inner ear and in the retina, which is the light-sensitive tissue at the back of the eye. In the inner ear, myosin VIIA plays a role in the development and maintenance of hairlike projections called stereocilia. Stereocilia, which are rich in actin, line the inner ear and bend in response to sound waves. This bending motion is critical for converting sound waves to nerve impulses, which are then transmitted to the brain. Stereocilia are also elements of the vestibular system, the part of the inner ear that helps maintain the body's balance and orientation in space. Bending of these stereocilia is needed to transmit signals from the vestibular system to the brain. In the retina, myosin VIIA is found primarily in a thin layer of cells called the retinal pigment epithelium (RPE). Myosin VIIA probably plays a role in the development and maintenance of this tissue, which supports and nourishes the retina. Research suggests that one function of myosin VIIA is to carry small sacs of pigment (called melanosomes) within the RPE. This pigment is necessary for normal vision. Myosin VIIA is also found in other parts of the retina, where it likely carries additional proteins and molecules that are important for vision. -

Head Region Mutations
Copyright 1998 by the Genetics Society of America Molecular Genetic Dissection of Mouse Unconventional Myosin-VA: Head Region Mutations Jian-Dong Huang,* M. Jamie T. V. Cope,²,1 Valerie Mermall,³ Marjorie C. Strobel,* John Kendrick-Jones,² Liane B. Russell,²² Mark S. Mooseker,³,§,** Neal G. Copeland,* and Nancy A. Jenkins,* *ABL-Basic Research Program, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, Maryland 21702, ²MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, United Kingdom, ³Department of Biology, §Department of Pathology, **Department of Cell Biology, Yale University, New Haven, Connecticut 06520, ²²Biology Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831 Manuscript received September 19, 1997 Accepted for publication December 23, 1997 ABSTRACT The mouse dilute (d) locus encodes unconventional myosin-VA (MyoVA). Mice carrying null alleles of dilute have a lightened coat color and die from a neurological disorder resembling ataxia and opisthotonus within three weeks of birth. Immunological and ultrastructural studies suggest that MyoVA is involved in the transport of melanosomes in melanocytes and smooth endoplasmic reticulum in cerebellar Purkinje cells. In studies described here, we have used an RT-PCR-based sequencing approach to identify the mutations responsible for 17 viable dilute alleles that vary in their effects on coat color and the nervous system. Seven of these mutations mapped to the MyoVA motor domain and are reported here. Crystallo- graphic modeling and mutant expression studies were used to predict how these mutations might affect motor domain function and to attempt to correlate these effects with the mutant phenotype. HE mouse dilute (d) locus encodes unconventional motor protein is used for the long-range transport of Tmyosin-VA (MyoVA). -

Novel Myosin Mutations for Hereditary Hearing Loss Revealed by Targeted Genomic Capture and Massively Parallel Sequencing
European Journal of Human Genetics (2014) 22, 768–775 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg ARTICLE Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing Zippora Brownstein1,6, Amal Abu-Rayyan2,6, Daphne Karfunkel-Doron1, Serena Sirigu3, Bella Davidov4, Mordechai Shohat1,4, Moshe Frydman1,5, Anne Houdusse3, Moien Kanaan2 and Karen B Avraham*,1 Hereditary hearing loss is genetically heterogeneous, with a large number of genes and mutations contributing to this sensory, often monogenic, disease. This number, as well as large size, precludes comprehensive genetic diagnosis of all known deafness genes. A combination of targeted genomic capture and massively parallel sequencing (MPS), also referred to as next-generation sequencing, was applied to determine the deafness-causing genes in hearing-impaired individuals from Israeli Jewish and Palestinian Arab families. Among the mutations detected, we identified nine novel mutations in the genes encoding myosin VI, myosin VIIA and myosin XVA, doubling the number of myosin mutations in the Middle East. Myosin VI mutations were identified in this population for the first time. Modeling of the mutations provided predicted mechanisms for the damage they inflict in the molecular motors, leading to impaired function and thus deafness. The myosin mutations span all regions of these molecular motors, leading to a wide range of hearing phenotypes, reinforcing the key role of this family of proteins in auditory function. This study demonstrates that multiple mutations responsible for hearing loss can be identified in a relatively straightforward manner by targeted-gene MPS technology and concludes that this is the optimal genetic diagnostic approach for identification of mutations responsible for hearing loss. -

A Mouse Forward Genetics Screen Identifies LISTERIN As an E3
A mouse forward genetics screen identifies INAUGURAL ARTICLE LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration Jessie Chua,1, Nancy A. Hongb,2, Claudio A. Masudac,3, Brian V. Jenkinsa, Keats A. Nelmsd, Christopher C. Goodnowd, Richard J. Glynnec, Hua Wub,4, Eliezer Masliahe, Claudio A. P. Joazeiroc,5, and Steve A. Kaya,6,7 aDepartment of Biochemistry, Institute for Childhood and Neglected Diseases, The Scripps Research Institute, ICND216, 10550 North Torrey Pines Road, La Jolla, CA 90237; bPhenomix Corporation, 5871 Oberlin Drive, Suite 200, San Diego, CA 92121; cGenomics Institute of the Novartis Research Foundation, 10675 John Jay Hopkins Drive, San Diego, CA 92121; dPhenomix Australia, Pty., Ltd., Level 3 Building 117, Australian Phenomics Facility, Garran Road, Acton, ACT 2601, Australia; and eDepartment of Neurosciences, University of California San Diego, School of Medicine, La Jolla, CA 92093 This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected in 2008. Contributed by Steve A. Kay, December 19, 2008 (sent for review November 13, 2008) A mouse neurological mutant, lister, was identified through a ENU generated a rat model for human Usher syndrome type 1B genome-wide N-ethyl-N-nitrosourea (ENU) mutagenesis screen. (9). Further, ENU-induced mutation in dynein led to progressive Homozygous lister mice exhibit profound early-onset and progres- motor neuron degeneration in mice (10). Although no human sive neurological and motor dysfunction. lister encodes a RING disease has yet been mapped to dynein itself, mutation in the finger protein, LISTERIN, which functions as an E3 ubiquitin ligase dynein activator, dynactin, causes degeneration of lower motor in vitro. -

LMO7 Deficiency Reveals the Significance of the Cuticular Plate For
ARTICLE https://doi.org/10.1038/s41467-019-09074-4 OPEN LMO7 deficiency reveals the significance of the cuticular plate for hearing function Ting-Ting Du1, James B. Dewey2, Elizabeth L. Wagner1, Runjia Cui3, Jinho Heo4, Jeong-Jin Park5, Shimon P. Francis1, Edward Perez-Reyes6, Stacey J. Guillot7, Nicholas E. Sherman5, Wenhao Xu8, John S Oghalai2, Bechara Kachar3 & Jung-Bum Shin1 Sensory hair cells, the mechanoreceptors of the auditory and vestibular systems, harbor 1234567890():,; two specialized elaborations of the apical surface, the hair bundle and the cuticular plate. In contrast to the extensively studied mechanosensory hair bundle, the cuticular plate is not as well understood. It is believed to provide a rigid foundation for stereocilia motion, but specifics about its function, especially the significance of its integrity for long-term maintenance of hair cell mechanotransduction, are not known. We discovered that a hair cell protein called LIM only protein 7 (LMO7) is specifically localized in the cuticular plate and the cell junction. Lmo7 KO mice suffer multiple cuticular plate deficiencies, including reduced filamentous actin density and abnormal stereociliar rootlets. In addition to the cuticular plate defects, older Lmo7 KO mice develop abnormalities in inner hair cell stereocilia. Together, these defects affect cochlear tuning and sensitivity and give rise to late-onset progressive hearing loss. 1 Department of Neuroscience, University of Virginia, Charlottesville, VA 22908, USA. 2 Caruso Department of Otolaryngology-Head and Neck Surgery, University of Southern California, Los Angeles, CA 90033, USA. 3 National Institute for Deafness and Communications Disorders, National Institute of Health, Bethesda, MD 20892, USA. 4 Center for Cell Signaling and Department of Microbiology, Immunology and Cancer Biology, University of Virginia, Charlottesville, VA 22908, USA. -
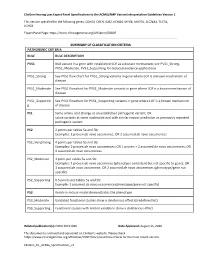
Clingen Hearing Loss Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1
ClinGen Hearing Loss Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1 This version specified for the following genes: CDH23, COCH, GJB2, KCNQ4, MYO6, MYO7A, SLC26A4, TECTA, USH2A Expert Panel Page: https://www.clinicalgenome.org/affiliation/50007 SUMMARY OF CLASSIFICATION CRITERIA PATHOGENIC CRITERIA RULE RULE DESCRIPTION PVS1 Null variant in a gene with established LOF as a disease mechanism; see PVS1_Strong, PVS1_Moderate, PVS1_Supporting for reduced evidence applications PVS1_Strong See PVS1 flow chart for PVS1_Strong variants in gene where LOF is a known mechanism of disease PVS1_Moderate See PVS1 flowchart for PVS1_Moderate variants in gene where LOF is a known mechanism of disease PVS1_Supportin See PVS1 flowchart for PVS1_Supporting variants in gene where LOF is a known mechanism g of disease PS1 Same amino acid change as an established pathogenic variant; OR splice variants at same nucleotide and with similar impact prediction as previously reported pathogenic variant PS2 2 points per tables 5a and 5b: Examples: 1 proven de novo occurrence; OR 2 assumed de novo occurrences PS2_VeryStrong 4 points per tables 5a and 5b: Examples: 2 proven de novo occurrences; OR 1 proven + 2 assumed de novo occurrences; OR 4 assumed de novo occurrences PS2_Moderate 1 point per tables 5a and 5b: Examples: 1 proven de novo occurrence (phenotype consistent but not specific to gene); OR 1 assumed de novo occurrence; OR 2 assumed de novo occurrences (phenotype/gene not specific) PS2_Supporting 0.5 points per tables 5a and 5b: Example: 1 assumed de novo occurrence (phenotype/gene not specific) PS3 Knock-in mouse model demonstrates the phenotype PS3_Moderate Validated functional studies show a deleterious effect (predefined list) PS3_Supporting Functional studies with limited validation show a deleterious effect Related publication(s): PMID 30311386 Date Approved: August 15, 2018 This document is archived and versioned on ClinGen’s website. -

Lamin B1 Is Required for Mature Neuron-Specific Gene Expression
ARTICLE Received 4 Jul 2016 | Accepted 28 Feb 2017 | Published 20 Apr 2017 DOI: 10.1038/ncomms15098 OPEN Lamin B1 is required for mature neuron-specific gene expression during olfactory sensory neuron differentiation Crystal M. Gigante1,2, Michele Dibattista3,4, Frederick N. Dong1, Xiaobin Zheng2, Sibiao Yue2, Stephen G. Young5, Johannes Reisert3, Yixian Zheng2 & Haiqing Zhao1 B-type lamins are major constituents of the nuclear lamina in all metazoan cells, yet have specific roles in the development of certain cell types. Although they are speculated to regulate gene expression in developmental contexts, a direct link between B-type lamins and developmental gene expression in an in vivo system is currently lacking. Here, we identify lamin B1 as a key regulator of gene expression required for the formation of functional olfactory sensory neurons. By using targeted knockout in olfactory epithelial stem cells in adult mice, we show that lamin B1 deficient neurons exhibit attenuated response to odour stimulation. This deficit can be explained by decreased expression of genes involved in mature neuron function, along with increased expression of genes atypical of the olfactory lineage. These results support that the broadly expressed lamin B1 regulates expression of a subset of genes involved in the differentiation of a specific cell type. 1 Department of Biology, The Johns Hopkins University, Baltimore, Maryland 21218, USA. 2 Department of Embryology, Carnegie Institution for Science, Baltimore, Maryland 21218, USA. 3 Monell Chemical Senses Center, Philadelphia, Pennsylvania 19104, USA. 4 Department of Basic Medical Sciences, Neuroscience and Sensory Organs, University of Bari ‘A. Moro’, Bari 70121, Italy. 5 Department of Medicine, Molecular Biology Institute and Department of Human Genetics, University of California, Los Angeles, California 90095, USA. -

Wnt Signaling Induces Proliferation of Sensory Precursors in the Postnatal Mouse Cochlea
Wnt signaling induces proliferation of sensory precursors in the postnatal mouse cochlea Renjie Chaia,1, Bryan Kuob,1, Tian Wanga, Eric J. Liawa, Anping Xiaa, Taha A. Jana,c, Zhiyong Liub, Makoto M. Taketod, John S. Oghalaia, Roeland Nussec,e,2, Jian Zuob, and Alan G. Chenga,2 Departments of aOtolaryngology-Head and Neck Surgery and eDevelopmental Biology and cHoward Hughes Medical Institute, Stanford University School of Medicine, Stanford, CA 94305; bDepartment of Developmental Neurobiology, St. Jude Children’s Research Hospital, Memphis, TN, 38103; and dDepartment of Pharmacology, Graduate School of Medicine, Kyoto University, Kyoto 606-8501, Japan Contributed by Roeland Nusse, April 9, 2012 (sent for review February 20, 2012) Inner ear hair cells are specialized sensory cells essential for found that these cells exhibit such progenitor behavior. These auditory function. Previous studies have shown that the sensory results corroborated with those of White et al. (7), who isolated epithelium is postmitotic, but it harbors cells that can behave as supporting cells using the p27(Kip1)-GFP mouse line. When progenitor cells in vitro, including the ability to form new hair p75-nerve growth factor receptor was used as an enrichment cells. Lgr5, a Wnt target gene, marks distinct supporting cell types marker, hair cell formation increased, raising the possibility that in the neonatal cochlea. Here, we tested the hypothesis that distinct supporting cell types are more capable of differentiating Lgr5+ cells are Wnt-responsive sensory precursor cells. In contrast into hair cells in vitro. To date, the in vivo behavior of cochlear to their quiescent in vivo behavior, Lgr5+ cells isolated by flow supporting cells in the postnatal period and the role of Wnt cytometry from neonatal Lgr5EGFP-CreERT2/+ mice proliferated and signaling in regulating their behavior remain unclear. -

(MYO7A) Auditory Mutation V a Street, J C Kallman, K L Kiemele
1of8 ELECTRONIC LETTER J Med Genet: first published as 10.1136/jmg.2003.013557 on 30 April 2004. Downloaded from Modifier controls severity of a novel dominant low- frequency MyosinVIIA (MYO7A) auditory mutation V A Street, J C Kallman, K L Kiemele ............................................................................................................................... J Med Genet 2004;41:e62 (http://www.jmedgenet.com/cgi/content/full/41/5/e62). doi: 10.1136/jmg.2003.013557 earing impairment is a common sensory deficit with both genetic and environmental aetiologies. Pre-lingual Key points Hhearing loss affects approximately 1 in every 1000 children in the United States with a genetic basis in about N Mutations within myosin molecules can lead to 50% of the cases.1 An additional 1 per 1000 individuals syndromic and non-syndromic hearing impairment. 1 experience auditory deficits prior to adulthood. Large N We describe the genetic mapping of progressive pedigrees with monogenic non-syndromic hearing impair- sensorineural hearing loss first affecting low-frequency ment have allowed genetic mapping of at least 80 chromo- auditory thresholds within a large human pedigree to somal locations harbouring auditory-related deafness (DFN) chromosome 11q13.5. A maximal pairwise LOD score loci with the identification of over 30 DFN genes.2 The DFN of 7.23 was obtained with marker D11S4207. inheritance pattern is designated by A dominant, B recessive, and M modifier, with a number following A, B,orM N WeidentifiedamyosinVIIA(MYO7A) G2164C indicating the relative order in which the locus was mutation that co-segregates with auditory dysfunction identified. For example, DFNA1 represents the first dom- in the pedigree. The mutation results in a predicted inantly inherited deafness locus mapped in humans.