Adaptive Clinical Trial Designs with Surrogates: When Should We Bother?*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Challenging Issues in Clinical Trial Design: Part 4 of a 4-Part Series on Statistics for Clinical Trials
Challenging Issues in Clinical Trial Design: Part 4 of a 4-part Series on Statistics for Clinical Trials Brief title: Challenges in Trial Design Stuart J. Pocock, PHD,* Tim C. Clayton, MSC,* Gregg W. Stone, MD† From the: *London School of Hygiene and Tropical Medicine, London, United Kingdom; †Columbia University Medical Center, New York-Presbyterian Hospital and the Cardiovascular Research Foundation, New York, New York <COR> Reprint requests and correspondence: Prof. Stuart J. Pocock, Department of Medical Statistics, London School of Hygiene and Tropical Medicine, Keppel Street, London, WC1E 7HT, United Kingdom Telephone: +44 20 7927 2413 Fax: +44 20 7637 2853 E-mail: [email protected] Disclosures: The authors declare no conflicts of interest for this paper. 1 Abstract As a sequel to last week’s article on the fundamentals of clinical trial design, this article tackles related controversial issues: noninferiority trials; the value of factorial designs; the importance and challenges of strategy trials; Data Monitoring Committees (including when to stop a trial early); and the role of adaptive designs. All topics are illustrated by relevant examples from cardiology trials. <KW>Key words: Noninferiority trials; Factorial designs; Strategy trials, Data Monitoring Committees; Statistical stopping guidelines; Adaptive designs; Randomized Controlled Trials As Topic; Abbreviations ACS = acute coronary syndrome CABG = coronary artery bypass graft CI = confidence interval CV = cardiovascular DMC = Data Monitoring Committee FDA = Food and Drug Administration MACE = major adverse cardiovascular event OMT = optimal medical therapy PCI = percutaneous coronary intervention 2 Introduction Randomized controlled trials are the cornerstone of clinical guidelines informing best therapeutic practices, however their design and interpretation may be complex and nuanced. -

Adaptive Clinical Trials: an Introduction
Adaptive clinical trials: an introduction What are the advantages and disadvantages of adaptive clinical trial designs? How and why were Introduction adaptive clinical Adaptive clinical trial design is trials developed? becoming a hot topic in healthcare research, with some researchers In 2004, the FDA published a report arguing that adaptive trials have the on the problems faced by the potential to get new drugs to market scientific community in developing quicker. In this article, we explain new medical treatments.2 The what adaptive trials are and why they report highlighted that the pace of were developed, and we explore both innovation in biomedical science is the advantages of adaptive designs outstripping the rate of advances and the concerns being raised by in the available technologies and some in the healthcare community. tools for evaluating new treatments. Outdated tools are being used to assess new treatments and there What are adaptive is a critical need to improve the effectiveness and efficiency of clinical trials? clinical trials.2 The current process for developing Adaptive clinical trials enable new treatments is expensive, takes researchers to change an aspect a long time and in some cases, the of a trial design at an interim development process has to be assessment, while controlling stopped after significant amounts the rate of type 1 errors.1 Interim of time and resources have been assessments can help to determine invested.2 In 2006, the FDA published whether a trial design is the most a “Critical Path Opportunities -

FDA Oncology Experience in Innovative Adaptive Trial Designs
Innovative Adaptive Trial Designs Rajeshwari Sridhara, Ph.D. Director, Division of Biometrics V Office of Biostatistics, CDER, FDA 9/3/2015 Sridhara - Ovarian cancer workshop 1 Fixed Sample Designs • Patient population, disease assessments, treatment, sample size, hypothesis to be tested, primary outcome measure - all fixed • No change in the design features during the study Adaptive Designs • A study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses based on analysis of data (interim data) from subjects in the study 9/3/2015 Sridhara - Ovarian cancer workshop 2 Bayesian Designs • In the Bayesian paradigm, the parameter measuring treatment effect is regarded as a random variable • Bayesian inference is based on the posterior distribution (Bayes’ Rule – updated based on observed data) – Outcome adaptive • By definition adaptive design 9/3/2015 Sridhara - Ovarian cancer workshop 3 Adaptive Designs (Frequentist or Bayesian) • Allows for planned design modifications • Modifications based on data accrued in the trial up to the interim time • Unblinded or blinded interim results • Control probability of false positive rate for multiple options • Control operational bias • Assumes independent increments of information 9/3/2015 Sridhara - Ovarian cancer workshop 4 Enrichment Designs – Prognostic or Predictive • Untargeted or All comers design: – post-hoc enrichment, prospective-retrospective designs – Marker evaluation after randomization (example: KRAS in cetuximab -

Survival Analysis: Part I — Analysis Korean Journal of Anesthesiology of Time-To-Event
Statistical Round pISSN 2005-6419 • eISSN 2005-7563 KJA Survival analysis: Part I — analysis Korean Journal of Anesthesiology of time-to-event Junyong In1 and Dong Kyu Lee2 Department of Anesthesiology and Pain Medicine, 1Dongguk University Ilsan Hospital, Goyang, 2Guro Hospital, Korea University School of Medicine, Seoul, Korea Length of time is a variable often encountered during data analysis. Survival analysis provides simple, intuitive results concerning time-to-event for events of interest, which are not confined to death. This review introduces methods of ana- lyzing time-to-event. The Kaplan-Meier survival analysis, log-rank test, and Cox proportional hazards regression model- ing method are described with examples of hypothetical data. Keywords: Censored data; Cox regression; Hazard ratio; Kaplan-Meier method; Log-rank test; Medical statistics; Power analysis; Proportional hazards; Sample size; Survival analysis. Introduction mation of the time [2]. The Korean Journal of Anesthesiology has thus far published In a clinical trial or clinical study, an investigational treat- several papers using survival analysis for clinical outcomes: a ment is administered to subjects, and the resulting outcome data comparison of 90-day survival rate for pressure ulcer develop- are collected and analyzed after a certain period of time. Most ment or non-development after surgery under general anesthesia statistical analysis methods do not include the length of time [3], a comparison of postoperative 5-year recurrence rate in as a variable, and analysis is made only on the trial outcomes patients with breast cancer depending on the type of anesthetic upon completion of the study period, as specified in the study agent used [4], and a comparison of airway intubation success protocol. -

FDA Comments on CBD in Foods
Popular Content Public Health Focus FDA and Marijuana: Questions and Answers 1. How is marijuana therapy being used by some members of the medical community? 2. Why hasn’t the FDA approved marijuana for medical uses? 3. Is marijuana safe for medical use? 4. How does FDA’s role differ from NIH and DEA’s role when it comes to the investigation of marijuana for medical use? 5. Does the FDA object to the clinical investigation of marijuana for medical use? 6. What kind of research is the FDA reviewing when it comes to the efficacy of marijuana? 7. How can patients get into expanded access program for marijuana for medical use? 8. Does the FDA have concerns about administering a cannabis product to children? 9. What is FDA’s reaction to states that are allowing marijuana to be sold for medical uses without the FDA’s approval? 10. What is the FDA’s position on state “Right to Try” bills? 11. Has the agency received any adverse event reports associated with marijuana for medical conditions? 12. Can products that contain cannabidiol be sold as dietary supplements? 13. Is it legal, in interstate commerce, to sell a food to which cannabidiol has been added? 14. In making the two previous determinations, why did FDA conclude that substantial clinical investigations have been authorized for and/or instituted about cannabidiol, and that the existence of such investigations has been made public? 15. Will FDA take enforcement action regarding cannabidiol products that are marketed as dietary supplements? What about foods to which cannabidiol has been added? 16. -
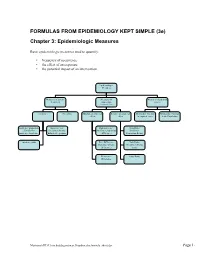
FORMULAS from EPIDEMIOLOGY KEPT SIMPLE (3E) Chapter 3: Epidemiologic Measures
FORMULAS FROM EPIDEMIOLOGY KEPT SIMPLE (3e) Chapter 3: Epidemiologic Measures Basic epidemiologic measures used to quantify: • frequency of occurrence • the effect of an exposure • the potential impact of an intervention. Epidemiologic Measures Measures of disease Measures of Measures of potential frequency association impact (“Measures of Effect”) Incidence Prevalence Absolute measures of Relative measures of Attributable Fraction Attributable Fraction effect effect in exposed cases in the Population Incidence proportion Incidence rate Risk difference Risk Ratio (Cumulative (incidence density, (Incidence proportion (Incidence Incidence, Incidence hazard rate, person- difference) Proportion Ratio) Risk) time rate) Incidence odds Rate Difference Rate Ratio (Incidence density (Incidence density difference) ratio) Prevalence Odds Ratio Difference Macintosh HD:Users:buddygerstman:Dropbox:eks:formula_sheet.doc Page 1 of 7 3.1 Measures of Disease Frequency No. of onsets Incidence Proportion = No. at risk at beginning of follow-up • Also called risk, average risk, and cumulative incidence. • Can be measured in cohorts (closed populations) only. • Requires follow-up of individuals. No. of onsets Incidence Rate = ∑person-time • Also called incidence density and average hazard. • When disease is rare (incidence proportion < 5%), incidence rate ≈ incidence proportion. • In cohorts (closed populations), it is best to sum individual person-time longitudinally. It can also be estimated as Σperson-time ≈ (average population size) × (duration of follow-up). Actuarial adjustments may be needed when the disease outcome is not rare. • In an open populations, Σperson-time ≈ (average population size) × (duration of follow-up). Examples of incidence rates in open populations include: births Crude birth rate (per m) = × m mid-year population size deaths Crude mortality rate (per m) = × m mid-year population size deaths < 1 year of age Infant mortality rate (per m) = × m live births No. -

Facilitating the Use of Imaging Biomarkers in Therapeutic Clinical
Facilitating the Use of Imaging Biomarkers in Therapeutic Clinical Trials Michael Graham, PhD, MD President, SNM Co-chair, Clinical Trials Network Facilitating the Use of Imaging Biomarkers in Therapeutic Clinical Trials • Definitions – Biomarker, Surrogate Biomarker • Standardization • Harmonization • Elements of a clinical trial • What can be facilitated • SNM Clinical Trials Network Imaging Biomarkers A biomarker is a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention. (FDA website) • Utility of imaging biomarkers in clinical trials – Assessing response to therapy (surrogate end point) • FDG • FLT – Stratifying patient populations • Receptor status (FES, SRS, etc.) • Hypoxia Surrogate Endpoints in Clinical Trials A surrogate endpoint is expected to predict clinical benefit (or harm, or lack of benefit) based on epidemiologic, therapeutic, pathophysiologic or other scientific evidence. (FDA website) • Assessing response to therapy – Relatively early “go vs. no go” decisions in Phase I or II – Decision point in adaptive designed trials – Building evidence for “validation” or “qualification” • Personalized medicine – Early identification of responders and non-responders Sohn HJ, et al. FLT PET before and 7 days after gefitinib (EGFR inhibitor) treatment predicts response in patients with advanced adenocarcinoma of the lung. Clin Cancer Res. 2008 Nov 15;14(22):7423-9. Imaging at 1 hr p 15 mCi FLT Threshold: -

Incidence and Secondary Transmission of SARS-Cov-2 Infections in Schools
Prepublication Release Incidence and Secondary Transmission of SARS-CoV-2 Infections in Schools Kanecia O. Zimmerman, MD; Ibukunoluwa C. Akinboyo, MD; M. Alan Brookhart, PhD; Angelique E. Boutzoukas, MD; Kathleen McGann, MD; Michael J. Smith, MD, MSCE; Gabriela Maradiaga Panayotti, MD; Sarah C. Armstrong, MD; Helen Bristow, MPH; Donna Parker, MPH; Sabrina Zadrozny, PhD; David J. Weber, MD, MPH; Daniel K. Benjamin, Jr., MD, PhD; for The ABC Science Collaborative DOI: 10.1542/peds.2020-048090 Journal: Pediatrics Article Type: Regular Article Citation: Zimmerman KO, Akinboyo IC, Brookhart A, et al. Incidence and secondary transmission of SARS-CoV-2 infections in schools. Pediatrics. 2021; doi: 10.1542/peds.2020- 048090 This is a prepublication version of an article that has undergone peer review and been accepted for publication but is not the final version of record. This paper may be cited using the DOI and date of access. This paper may contain information that has errors in facts, figures, and statements, and will be corrected in the final published version. The journal is providing an early version of this article to expedite access to this information. The American Academy of Pediatrics, the editors, and authors are not responsible for inaccurate information and data described in this version. Downloaded from©2021 www.aappublications.org/news American Academy by of guest Pediatrics on September 27, 2021 Prepublication Release Incidence and Secondary Transmission of SARS-CoV-2 Infections in Schools Kanecia O. Zimmerman, MD1,2,3; Ibukunoluwa C. Akinboyo, MD1,2; M. Alan Brookhart, PhD4; Angelique E. Boutzoukas, MD1,2; Kathleen McGann, MD2; Michael J. -

Evolution of Clinical Trials Throughout History
Evolution of clinical trials throughout history Emma M. Nellhaus1, Todd H. Davies, PhD1 Author Affiliations: 1. Office of Research and Graduate Education, Marshall University Joan C. Edwards School of Medicine, Huntington, West Virginia The authors have no financial disclosures to declare and no conflicts of interest to report. Corresponding Author: Todd H. Davies, PhD Director of Research Development and Translation Marshall University Joan C. Edwards School of Medicine Huntington, West Virginia Email: [email protected] Abstract The history of clinical research accounts for the high ethical, scientific, and regulatory standards represented in current practice. In this review, we aim to describe the advances that grew from failures and provide a comprehensive view of how the current gold standard of clinical practice was born. This discussion of the evolution of clinical trials considers the length of time and efforts that were made in order to designate the primary objective, which is providing improved care for our patients. A gradual, historic progression of scientific methods such as comparison of interventions, randomization, blinding, and placebos in clinical trials demonstrates how these techniques are collectively responsible for a continuous advancement of clinical care. Developments over the years have been ethical as well as clinical. The Belmont Report, which many investigators lack appreciation for due to time constraints, represents the pinnacle of ethical standards and was developed due to significant misconduct. Understanding the history of clinical research may help investigators value the responsibility of conducting human subjects’ research. Keywords Clinical Trials, Clinical Research, History, Belmont Report In modern medicine, the clinical trial is the gold standard and most dominant form of clinical research. -

Surrogate Endpoint Biomarkers for Cervical Cancer Chemoprevention Trials
Journal of Cellular Biochemistry, Supplement 23:113-124 (1 995) Surrogate Endpoint Biomarkers for Cervical Cancer Chemoprevention Trials Mack T. Ruffin IV, MD, MPH: Mohammed S. Qgaily, MD: Carolyn M. Johnston, MD? Lucie Gregoire, PhD: Wayne D. Lancaster, PhD; and Dean E. Brenner, MD6 Department of Family Practice, University of Michigan Medical Center, Ann Arbor, MI 48109-0708 Department of Internal Medicine, Division of Hematology and Oncology, Simpson Memorial Institute, Ann Arbor, MI 48109-0724 Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, Ann Arbor, MI 48109-0718 Department of Obstetrics and Gynecology, Wayne State University School of Medicine, Detroit, MI 48201 Department of Obstetrics and Gynecology, Center for Molecular Medicine and Genetics, Detroit, MI 48201 Department of Internal Medicine, Division of Hematology and Oncology, Simpson Memorial Institute, Ann Arbor, MI 48109-0724 Abstract Cervical intraepithelial neoplasia (CIN) represents a spectrum of epithelial changes that provide an excellent model for developing chemopreventive interventions for cervical cancer. Possible drug effect surrogate endpoint biomarkers are dependent on the agent under investigation. Published and preliminary clinical reports suggest retinoids and carotenoids are effective chemopreventive agents for CIN. Determination of plasma and tissue pharmacology of these agents and their metabolites could serve as drug effect intermediate endpoints. In addition, retinoic acid receptors could serve as both drug and biological effect intermediate endpoints. Possible biological effect surrogate endpoint biomarkers include cytomorphological parameters, proliferation markers, genomic markers, regulatory markers, and differentiation. Given the demonstrated causality of human papillomavirus (HPV) for cervical cancer, establishing the relationship to HPV will be an essential component of any biological intermediate endpoint biomarker. -

Delineating Virulence of Vibrio Campbellii
www.nature.com/scientificreports OPEN Delineating virulence of Vibrio campbellii: a predominant luminescent bacterial pathogen in Indian shrimp hatcheries Sujeet Kumar1*, Chandra Bhushan Kumar1,2, Vidya Rajendran1, Nishawlini Abishaw1, P. S. Shyne Anand1, S. Kannapan1, Viswas K. Nagaleekar3, K. K. Vijayan1 & S. V. Alavandi1 Luminescent vibriosis is a major bacterial disease in shrimp hatcheries and causes up to 100% mortality in larval stages of penaeid shrimps. We investigated the virulence factors and genetic identity of 29 luminescent Vibrio isolates from Indian shrimp hatcheries and farms, which were earlier presumed as Vibrio harveyi. Haemolysin gene-based species-specifc multiplex PCR and phylogenetic analysis of rpoD and toxR identifed all the isolates as V. campbellii. The gene-specifc PCR revealed the presence of virulence markers involved in quorum sensing (luxM, luxS, cqsA), motility (faA, lafA), toxin (hly, chiA, serine protease, metalloprotease), and virulence regulators (toxR, luxR) in all the isolates. The deduced amino acid sequence analysis of virulence regulator ToxR suggested four variants, namely A123Q150 (AQ; 18.9%), P123Q150 (PQ; 54.1%), A123P150 (AP; 21.6%), and P123P150 (PP; 5.4% isolates) based on amino acid at 123rd (proline or alanine) and 150th (glutamine or proline) positions. A signifcantly higher level of the quorum-sensing signal, autoinducer-2 (AI-2, p = 2.2e−12), and signifcantly reduced protease activity (p = 1.6e−07) were recorded in AP variant, whereas an inverse trend was noticed in the Q150 variants AQ and PQ. The pathogenicity study in Penaeus (Litopenaeus) vannamei juveniles revealed that all the isolates of AQ were highly pathogenic with Cox proportional hazard ratio 15.1 to 32.4 compared to P150 variants; PP (5.4 to 6.3) or AP (7.3 to 14). -

U.S. Investments in Medical and Health Research and Development 2013 - 2017 Advocacy Has Helped Bring About Five Years of Much-Needed Growth in U.S
Fall 2018 U.S. Investments in Medical and Health Research and Development 2013 - 2017 Advocacy has helped bring about five years of much-needed growth in U.S. medical and health research investment. More advocacy is critical now to ensure our nation steps up in response to health threats that we can—we must—overcome. More Than Half Favor Doubling Federal Spending on Medical Research Do you favor or oppose doubling federal spending on medical research over the next five years? 19% Not Sure 23% 8% Strongly favor Strongly oppose 16% 35% Somewhat oppose Somewhat favor Source: A Research!America survey of U.S. adults conducted in partnership with Zogby Analytics in January 2018. Research!America 3 Introduction Investment1 in medical and health research and development (R&D) in the U.S. grew by $38.8 billion or 27% from 2013 to 2017. Industry continues to invest more than any other sector, accounting for 67% of total spending in 2017, followed by the federal government at 22%. Federal investments increased from 2016 to 2017, the second year of growth after a dip from 2014 to 2015. Overall, federal investment increased by $6.1 billion or 18.4% from 2013 to 2017, but growth has been uneven across federal health agencies. Investment by other sectors, including academic and research institutions, foundations, state and local governments, and voluntary health associations and professional societies, also increased from 2013 to 2017. Looking ahead, medical and health R&D spending is expected to move in an upward trajectory in 2018 but will continue to fall short relative to the health and economic impact of major health threats.