Online Supplement
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Publication a Bypass and HEWS
Hospital Bypass and Hospital Early Warning System July to December 2010 Department of Health Hospital Bypass and Hospital Early Warning System July to December 2010 Page 1 Hospital Bypass and HEWS (July – December 2010) If you would like to receive this publication in an accessible format, please phone (03) 9096 7392 using the National Relay service 13 36 77 if required. © Copyright, State of Victoria, Department of Health, 2011 Published by the Hospital and Health Service Performance Division, Victorian Government, Department of Health, Melbourne, Victoria. This publication is copyright, no part may be reproduced by any process except in accordance with the provisions of the Copyright Act 1968. This document is also available in PDF format on the internet at: www.health.vic.gov.au/performance Authorised by the State Government of Victoria, 50 Lonsdale Street, Melbourne. Page 2 Contents Hospital Bypass and Hospital Early Warning System (HEWS) 4 Introduction 4 Hospital Bypass 5 Hospital Early Warning System 8 Definitions 11 Hospital Bypass Specifications 11 Page 3 Hospital Bypass and HEWS (July – December 2010) Hospital Bypass and Hospital Early Warning System (HEWS) July to December 2010 Introduction This is an interim report containing summary information relating to the Emergency Department activity of Victoria's public hospitals. In future months, a wider range of information will be reported and made available through a new and dedicated website designed to provide information about hospital activity and performance in Victoria. March 2011 Page 4 Hospital Bypass Hospital bypass is a period of time when a public hospital emergency department can request that ambulances bypass it and take patients to other hospitals. -

Metropolitan Melbourne Public Hospitals Hospitals Current As at 19 July 2006 Sunbury Private
The Northern Hospital Healesville & District Hospital Broadmeadows Health Service Bundoora Extended Care Centre Heidelberg Repatriation Hospital Austin Hospital and Sunshine Hospital Mercy Hospital for Women Royal Melbourne Hospital, Royal Park Western Hospital RCH Royal Talbot Rehablilitation Centre Tweedle Child & Family Health Centre RMH RWH Caritas Dental HS St George's Health Service Maroondah Hospital St Vincent's RVEEH Box Hill Hospital Peter MacCallum O'Connell Family Centre (Grey Sisters) Inc. The Alfred The Peter James Centre Williamstown Hospital Mercy Werribee Caulfield General Medical Centre Calvary Health Care Bethlehem Ltd Angliss Hospital Monash Medical Centre, Moorabbin Monash Medical Centre, Clayton Hampton Rehabilitation Hospital Sandringham & District Hospital Kingston Centre Queen Elizabeth Centre Dandenong Hospital Casey Hospital Royal Children's Hospital Dental Health Services Victoria Royal Melbourne Hospital Royal Women's Hospital Cranbourne Integrated Care Centre St Vincent's Hospital Caritas Christi Hospice Royal Victorian Eye & Ear Hospital Peter MacCallum Cancer Centre INSETINSET Frankston Hospital 0 500 1000 Metres Mt Eliza Rehabilitation, Aged and Palliative Care 01020 Kilometres Rosebud Hospital Metropolitan Melbourne public hospitals Hospitals current as at 19 July 2006 Sunbury Private Northpark Private Hospital Gambro Diamond Valley Clinic Victorian Rehabilitation Centre - Northern Melbourne Reservoir Private Hospital Essendon Private Hospital Victoria Warringal Private Hospital John Fawkner Private Hospital -

Postgraduate Medical Council of Victoria Inc
POSTGRADUATE MEDICAL COUNCIL OF VICTORIA INC. CONTENTS Albury Wodonga Health .......................................................................................................................... 3 Alfred Health ........................................................................................................................................... 3 Austin Health........................................................................................................................................... 4 Austin Health – COMBINED NURSING / MENTAL HEALTH .................................................................... 4 Barwon Health Services .......................................................................................................................... 5 Bendigo Health ........................................................................................................................................ 5 Eastern Health ......................................................................................................................................... 6 Eastern Health – COMBINED NURSING / MENTAL HEALTH ................................................................... 6 Goulburn Valley Health ........................................................................................................................... 7 Latrobe Regional Hospital - General/Mental Health Nursing Stream .................................................... 8 Mercy Health .......................................................................................................................................... -

Hospital Services Report
Hospital Services Report December quarter 2001 Notes This document contains the most up-to-date information available at the time of preparation. This Hospital Services Report is available on the Department of Human Services Internet site located at: http://www.health.vic.gov.au/hsr/index.htm Design and production by Department of Human Services, Melbourne, Victoria. December 2001 © Copyright Department of Human Services 2001. This publication is copyright. No part may be reproduced by any process except in accordance with the provisions of the Copyright Act 1968. ii Contents Introduction 1 1. Patients waiting for residential aged care placement 2 2. Hospital admitted patient activity 4 3. Access to emergency services 6 4. Access to critical care services 15 5. Access to elective surgery 17 6. Private health insurance 28 7. Glossary 29 iii Introduction A number of graphs and tables refer to major metropolitan hospitals. Consumers, health care providers and government all need information on the The major metropolitan hospitals include: quality of health services. Public access to information assists consumers to Angliss Hospital understand the health care system, it assists providers of health services in Austin & Repatriation Medical Centre planning and increases the accountability of the Department of Human - Austin campus Services to the people in the state of Victoria, Australia. - Repatriation campus Box Hill Hospital The Hospital Services Report was introduced in 1995. This edition includes Dandenong Hospital data for the December quarter 2001, which covers the months of October, Frankston Hospital November and December. Maroondah Hospital Monash Medical Centre The information included in this report is often requested from the Department. -

Parliament of Victoria Parliamentary Debates (Hansard)
PARLIAMENT OF VICTORIA PARLIAMENTARY DEBATES (HANSARD) LEGISLATIVE COUNCIL FIFTY-NINTH PARLIAMENT FIRST SESSION WEDNESDAY, 4 AUGUST 2021 hansard.parliament.vic.gov.au By authority of the Victorian Government Printer The Governor The Honourable LINDA DESSAU, AC The Lieutenant-Governor The Honourable KEN LAY, AO, APM The ministry Premier........................................................ The Hon. DM Andrews, MP Deputy Premier, Minister for Education and Minister for Mental Health .. The Hon. JA Merlino, MP Attorney-General and Minister for Emergency Services .............. The Hon. J Symes, MLC Minister for Transport Infrastructure and Minister for the Suburban Rail Loop ....................................................... The Hon. JM Allan, MP Minister for Training and Skills and Minister for Higher Education .... The Hon. GA Tierney, MLC Treasurer, Minister for Economic Development and Minister for Industrial Relations ........................................... The Hon. TH Pallas, MP Minister for Public Transport and Minister for Roads and Road Safety . The Hon. BA Carroll, MP Minister for Energy, Environment and Climate Change and Minister for Solar Homes ................................................ The Hon. L D’Ambrosio, MP Minister for Child Protection and Minister for Disability, Ageing and Carers ...................................................... The Hon. LA Donnellan, MP Minister for Health, Minister for Ambulance Services and Minister for Equality .................................................... The Hon. MP -

Hospital Services Report
Hospital Services Report September quarter 2002 Notes This document contains the most up-to-date information available at the time of preparation. This Hospital Services Report is available on the Department of Human Services Internet site located at: http://www.health.vic.gov.au/hsr/index.htm Design and production by Department of Human Services, Melbourne, Victoria. September 2002 © Copyright Department of Human Services 2002. This publication is copyright. No part may be reproduced by any process except in accordance with the provisions of the Copyright Act 1968. ii Contents Introduction 1 1. Patients waiting for residential aged care placement 2 2. Hospital admitted patient activity 4 3. Access to emergency services 6 4. Access to critical care services 15 5. Access to elective surgery 17 6. Private health insurance 28 7. Glossary 29 iii Introduction Consumers, health care providers and government all need information on the Access to Emergency Services quality of health services. Public access to information assists consumers to From 1 January 2002, emergency department data in this report are derived from understand the health care system, it assists providers of health services in electronic rather than paper-based reporting. planning and increases the accountability of the Department of Human Services to the people in the state of Victoria, Australia. A number of graphs and tables refer to major metropolitan hospitals. The major metropolitan hospitals include: The Hospital Services Report was introduced in 1995. This edition includes data Angliss Hospital for the September quarter 2002, which covers the months of July, August and Austin & Repatriation Medical Centre September. -

Annual Report 2019 Connected Care 15
Every patient matters and we are committed to providing high-quality patient care, delivered by a skilled, dedicated, compassionate workforce. We continue to innovate and find new ways of caring, in line with community need. Continually improving our patients’ experience, on each and every touchpoint of their journey with us, is always our goal. Epworth HealthCare is Victoria’s largest not-for-profit private hospital group, renowned for excellence in diagnosis, treatment, care and rehabilitation. Epworth is an innovator in Australia’s health system, embracing the latest in evidence-based medicine to pioneer treatments and services for our patients. Our vision: Caring for people. Innovating for a healthy community. Our purpose: Every patient matters. We strive to improve health outcomes and experience through compassion, collaboration, learning and innovation. Our values: Respect, excellence, community, compassion, integrity, accountability. Epworth was founded in 1920 as a 25-bed community intermediate hospital in Richmond by a Methodist minister for those on moderate incomes. We will celebrate our centenary next year. Today, our care is world-class, our technology is state- of-the-art and our ethos remains focused on our patients. This Annual Report details achievements and highlights of the 2018–19 financial year. It follows the pillars of our Strategic Plan: Connected Care, Empowered People, Innovative Practice and Sustainability. This report is available online at epworth.org.au We also highlight some of our patient stories - as patients are at the heart of all we do. Acknowledgement of Country Epworth HealthCare acknowledges the people of the Kulin Nations, on whose land we work and care for our patients. -

An Analysis of the Victorian Rehabilitation and Recovery Care
An analysis of the Victorian rehabilitation and recovery care service system for people with severe mental illness and associated disability Project report 4 Clinical review of area mental health services 1997-2004 Intensive care for adults in Victorian public hospitals 2003 i An analysis of the Victorian rehabilitation and recovery care service system for people with severe mental illness and associated disability Project report ii Intensive care for adults in Victorian public hospitals 2003 Published by the Victorian Government Department of Human Services, Melbourne, Victoria © Copyright State of Victoria 2007 This publication is copyright, no part may be reproduced by any process except in accordance with the provisions of the Copyright Act 1968. This document may also be downloaded from the Department of Human Services website at: www.health.vic.gov.au/mental health/atoz.htm Authorised by the State Government of Victoria, 50 Lonsdale Street, Melbourne. August 2007 (070815) Contents 1 Introduction 1 2 Overview of relevant service system elements 5 3 Project methodology and data sources 15 4 How do the service elements currently operate? 17 5 Guiding principles for system development 33 6 Defining the issues 35 7 Proposed solutions 41 8 Recommendations 49 Appendix 1: Distribution of bed-based service elements 51 Appendix 2: Developing an integrated rehabilitation and care service system–flow chart 53 Common acronyms and initialisations used in this report AMHS area mental health service CAMHS child and adolescent mental health service -

The Royal Children's Hospital Melbourne
The Royal Children’s Hospital Melbourne Annual Financial Report 2010 – 11 On the move 2011 Opening in late 2011, the new Royal Children’s Hospital (RCH) will provide world class facilities befitting of our great hospital for children, families, visitors and staff. The move to and opening of the new hospital campus represents not only a milestone in the hospital’s history, but also in the history of paediatric care in this country and globally. Established in 1870, the hospital moved to its current site in Parkville in 1962 and was officially opened by Her Majesty, The Queen on February 26, 1963. With the building of our new hospital, we have taken the opportunity to create an environment that is uniquely designed for children and young people – creating spaces that are fun, stimulating and take healing beyond the bedside. The design reflects growing evidence of the importance of nature and environment to the healing process and there is an overwhelming sense of colour and light throughout the building, with beautiful artwork designed to appeal to and interest children of all ages. In the 48 years since the opening of this site there has been a significant expansion of not only the hospital’s clinical facilities but also teaching and research facilities, with our on-site campus partners, Murdoch Children’s Research Institute and The University of Melbourne Department of Paediatrics. Our expertise, leadership and reputation in paedatrics has grown, particularly in the treatment of childhood cancer, the surgery of congenital abnormalities, gastroenterology, cardiac surgery and epilepsy surgery. The RCH has touched the lives of children and families not just in this state but nationwide and internationally and in 2011, we join in celebration and recognition of our glorious past and with great excitement for the future. -

Professor of Cardiology Eastern Health Clinical School Faculty of Medicine, Nursing and Health Sciences Monash University
PROFESSOR OF CARDIOLOGY EASTERN HEALTH CLINICAL SCHOOL FACULTY OF MEDICINE, NURSING AND HEALTH SCIENCES MONASH UNIVERSITY AND DEPARTMENT OF CARDIOLOGY EASTERN HEALTH Position description Organisational context EASTERN HEALTH Our Vision Great health and wellbeing. Our Mission To provide positive health experiences for people and communities in the east. Organisational Environment Eastern Health provides a comprehensive range of high quality acute, sub-acute, palliative care, mental health, drug and alcohol, residential care and community health services to people and communities that are diverse in culture, age and socio-economic status. These services are provided both on an inpatient and outpatient basis. These services and programs are supported by staff who possess an unparalleled commitment to patient care as well as teaching and research. The strategic goals are fully detailed in the Eastern Health e-learning orientation. Eastern Health Values Eastern Health values reflect who we are as individuals and as an organisation, and are testament to the Eastern Health Mission Statement; they include the concepts of Excellence, Accountability, Compassion, Team Work, Integrity, Respect and Collaboration. The Eastern Health Values are fully detailed in the Code of Conduct which is covered in the Eastern Health e-learning orientation. Eastern Health Department of Cardiology The Eastern Health Department of Cardiology provides specialist care to Eastern Health patients with cardiac disease, providing a comprehensive range of interventional and non-interventional services. The Eastern Health Department of Cardiology is based at Box Hill Hospital. The Cardiac Catheter Laboratory is located at Box Hill Hospital, with interventional services provided across two laboratories with 14 recovery spaces servicing coronary and device/EP work. -
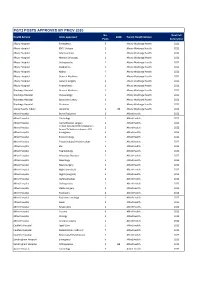
JMO POST DATA Current
PGY2 POSTS APPROVED BY PMCV 2020 No. Next Full Health Service Units approved 1008 Parent Health Service Posts Survey Due Albury Hospital Emergency 5 Albury Wodonga Health 2021 Albury Hospital ENT/ Urology 1 Albury Wodonga Health 2021 Albury Hospital Intensive Care 1 Albury Wodonga Health 2021 Albury Hospital Medical Oncology 1 Albury Wodonga Health 2021 Albury Hospital Orthopaedics 1 Albury Wodonga Health 2021 Albury Hospital Paediatrics 1 Albury Wodonga Health 2021 Albury Hospital Nights 4 Albury Wodonga Health 2021 Albury Hospital General Medicine 1 Albury Wodonga Health 2021 Albury Hospital General Surgery 2 Albury Wodonga Health 2021 Albury Hospital Anaesthetics 1 Albury Wodonga Health 2021 Wodonga Hospital General Medicine 1 Albury Wodonga Health 2021 Wodonga Hospital Gynaecology 1 Albury Wodonga Health 2021 Wodonga Hospital Specialist Surgery 1 Albury Wodonga Health 2021 Wodonga Hospital Geriatrics 1 Albury Wodonga Health 2021 Mercy Health, Albury Geriatrics 1 23 Albury Wodonga Health 2021 Alfred Hospital Burns/Faciomax 2 Alfred Health 2022 Alfred Hospital Cardiology 2 Alfred Health 2022 Alfred Hospital Cardiothoracic surgery 2 Alfred Health 2022 Critical Care (2x3mth rotations in Alfred Hospital 2 Alfred Health 2022 Anaes/ 2x3mth rotations in ICU) Alfred Hospital Emergency 4 Alfred Health 2022 Alfred Hospital Endocrinology 1 Alfred Health 2022 Alfred Hospital Endocrinology/ rheumatology 1 Alfred Health 2022 Alfred Hospital ENT 1 Alfred Health 2022 Alfred Hospital Haematology 1 Alfred Health 2022 Alfred Hospital Infectious Diseases -

Health Service Violence Prevention Fund Rounds 1, 2 and 3 Acute Health and Mental Health
Health Service Violence Prevention Fund Rounds 1, 2 and 3 Acute Health and Mental Health The Victorian Government has allocated $17 million in three rounds of funding for capital works to prevent violence in public health services. Funding has been allocated as follows: Round one: 2015–16 $3 million for violence prevention projects to health facilities, with half going to acute and half to mental health services. Round two: 2016–17 $7 million between acute and mental health services for violence prevention Round three: 2017–18 $7 million to health services for safety improvements in acute and mental health wards. Details of the projects are detailed in this document. We encourage members to report back to ANMF via anmfvic.asn.au/hsvpf on the status of projects in rounds 1, 2 and 3. anmfvic.asn.au/hsvpf 2015–16 Health Service Violence Prevention Fund Round 1 | Acute Health Region Health Service Hospital / Campus/ Project scope / replacement asset Community Health Centre Metropolitan Austin Health Austin Hospital – Emergency Fixed duress buttons to ambulance triage entry, CCTV in Department assessment area and upgrade of existing CCTV in central monitor at staff base. Djerriwarrh Health Bacchus Marsh Hospital – Installing a more direct route from the hospital to the car Services external area park. Installing lighting and security cameras. Bacchus Marsh Hospital Installing building access control to external doors and – Hospital Access Control some internal doors located in the main hospital building System and the administration building. Mercy Health Werribee Mercy Hospital Installation of CCTV, duress buttons, reel switches and (Outpatients Department) keypads to external and internal doors to facilitate controlled entry via swipe card, and addition of a press button at main foyer for general egress.