Necroinflammation in Kidney Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Signal Integration of Syk-Coupled C-Type Lectin Receptors
Contact, Collaboration, and Conflict: Signal Integration of Syk-Coupled C-Type Lectin Receptors This information is current as Jenny Ostrop and Roland Lang of September 27, 2021. J Immunol 2017; 198:1403-1414; ; doi: 10.4049/jimmunol.1601665 http://www.jimmunol.org/content/198/4/1403 Downloaded from References This article cites 202 articles, 75 of which you can access for free at: http://www.jimmunol.org/content/198/4/1403.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 27, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Th eJournal of Brief Reviews Immunology Contact, Collaboration, and Conflict: Signal Integration of Syk-Coupled C-Type Lectin Receptors Jenny Ostrop*,† and Roland Lang‡ Several spleen tyrosine kinase–coupled C-type lectin of TLRs and crosstalk of TLR signaling have been studied for receptors (CLRs) have emerged as important pattern almost two decades. -

Gene List HTG Edgeseq Immuno-Oncology Assay
Gene List HTG EdgeSeq Immuno-Oncology Assay Adhesion ADGRE5 CLEC4A CLEC7A IBSP ICAM4 ITGA5 ITGB1 L1CAM MBL2 SELE ALCAM CLEC4C DST ICAM1 ITGA1 ITGA6 ITGB2 LGALS1 MUC1 SVIL CDH1 CLEC5A EPCAM ICAM2 ITGA2 ITGAL ITGB3 LGALS3 NCAM1 THBS1 CDH5 CLEC6A FN1 ICAM3 ITGA4 ITGAM ITGB4 LGALS9 PVR THY1 Apoptosis APAF1 BCL2 BID CARD11 CASP10 CASP8 FADD NOD1 SSX1 TP53 TRAF3 BCL10 BCL2L1 BIRC5 CASP1 CASP3 DDX58 NLRP3 NOD2 TIMP1 TRAF2 TRAF6 B-Cell Function BLNK BTLA CD22 CD79A FAS FCER2 IKBKG PAX5 SLAMF1 SLAMF7 SPN BTK CD19 CD24 EBF4 FASLG IKBKB MS4A1 RAG1 SLAMF6 SPI1 Cell Cycle ABL1 ATF1 ATM BATF CCND1 CDK1 CDKN1B NCL RELA SSX1 TBX21 TP53 ABL2 ATF2 AXL BAX CCND3 CDKN1A EGR1 REL RELB TBK1 TIMP1 TTK Cell Signaling ADORA2A DUSP4 HES1 IGF2R LYN MAPK1 MUC1 NOTCH1 RIPK2 SMAD3 STAT5B AKT3 DUSP6 HES5 IKZF1 MAF MAPK11 MYC PIK3CD RNF4 SOCS1 STAT6 BCL6 ELK1 HEY1 IKZF2 MAP2K1 MAPK14 NFATC1 PIK3CG RORC SOCS3 SYK CEBPB EP300 HEY2 IKZF3 MAP2K2 MAPK3 NFATC3 POU2F2 RUNX1 SPINK5 TAL1 CIITA ETS1 HEYL JAK1 MAP2K4 MAPK8 NFATC4 PRKCD RUNX3 STAT1 TCF7 CREB1 FLT3 HMGB1 JAK2 MAP2K7 MAPKAPK2 NFKB1 PRKCE S100B STAT2 TYK2 CREB5 FOS HRAS JAK3 MAP3K1 MEF2C NFKB2 PTEN SEMA4D STAT3 CREBBP GATA3 IGF1R KIT MAP3K5 MTDH NFKBIA PYCARD SMAD2 STAT4 Chemokine CCL1 CCL16 CCL20 CCL25 CCL4 CCR2 CCR7 CX3CL1 CXCL12 CXCL3 CXCR1 CXCR6 CCL11 CCL17 CCL21 CCL26 CCL5 CCR3 CCR9 CX3CR1 CXCL13 CXCL5 CXCR2 MST1R CCL13 CCL18 CCL22 CCL27 CCL7 CCR4 CCRL2 CXCL1 CXCL14 CXCL6 CXCR3 PPBP CCL14 CCL19 CCL23 CCL28 CCL8 CCR5 CKLF CXCL10 CXCL16 CXCL8 CXCR4 XCL2 CCL15 CCL2 CCL24 CCL3 CCR1 CCR6 CMKLR1 CXCL11 CXCL2 CXCL9 CXCR5 -

C-Type Lectins in Veterinary Species: Recent Advancements and Applications
International Journal of Molecular Sciences Review C-Type Lectins in Veterinary Species: Recent Advancements and Applications Dimitri Leonid Lindenwald and Bernd Lepenies * Immunology Unit & Research Center for Emerging Infections and Zoonoses (RIZ), University for Veterinary Medicine Hannover, Foundation, 30559 Hannover, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-(0)5-11/953-6135 Received: 29 June 2020; Accepted: 17 July 2020; Published: 20 July 2020 Abstract: C-type lectins (CTLs), a superfamily of glycan-binding receptors, play a pivotal role in the host defense against pathogens and the maintenance of immune homeostasis of higher animals and humans. CTLs in innate immunity serve as pattern recognition receptors and often bind to glycan structures in damage- and pathogen-associated molecular patterns. While CTLs are found throughout the whole animal kingdom, their ligand specificities and downstream signaling have mainly been studied in humans and in model organisms such as mice. In this review, recent advancements in CTL research in veterinary species as well as potential applications of CTL targeting in veterinary medicine are outlined. Keywords: C-type lectin; glycans; immune modulation; comparative immunology; veterinary immunology 1. Introduction Glycans belong to the most abundant macromolecules constituting all living organisms. In multicellular animals, processes such as cell migration, homeostasis maintenance, and innate immune signaling rely on the ability of cells to recognize glycoconjugates, most often in the form of glycoproteins and glycolipids, via glycan binding proteins, the so-called lectins [1]. In the immune system, lectin receptors are either secreted or found on the cell surface of immune cells [2]. -

Beryllium-Induced Lung Disease Exhibits Expression Profiles Similar to Sarcoidosis
ORIGINAL ARTICLE BERYLLIUM-INDUCED LUNG DISEASE Beryllium-induced lung disease exhibits expression profiles similar to sarcoidosis Li Li1,2, Lori J. Silveira1, Nabeel Hamzeh1,2, May Gillespie1, Peggy M. Mroz1, Annyce S. Mayer1,2,3, Tasha E. Fingerlin1 and Lisa A. Maier1,2,3 Affiliations: 1Dept of Medicine, National Jewish Health, Denver, CO, USA. 2Division of Pulmonary and Critical Care Sciences, Dept of Medicine, School of Medicine, Denver, CO, USA. 3Environmental Occupational Health Dept, School of Public Health, University of Colorado, Denver, CO, USA. Correspondence: Li Li, Division of Environmental and Occupational Health Sciences, Dept of Medicine, National Jewish Health, 1400 Jackson Street, Denver, CO 80206, USA. E-mail: [email protected] ABSTRACT A subset of beryllium-exposed workers develop beryllium sensitisation (BeS) which precedes chronic beryllium disease (CBD). We conducted an in-depth analysis of differentially expressed candidate genes in CBD. We performed Affymetrix GeneChip 1.0 ST array analysis on peripheral blood mononuclear cells (PBMCs) from 10 CBD, 10 BeS and 10 beryllium-exposed, nondiseased controls stimulated with BeSO4 or medium. The differentially expressed genes were validated by high-throughput real-time PCR in this group and in an additional group of cases and nonexposed controls. The functional roles of the top candidate genes in CBD were assessed using a pharmacological inhibitor. CBD gene expression data were compared with whole blood and lung tissue in sarcoidosis from the Gene Expression Omnibus. We confirmed almost 450 genes that were significantly differentially expressed between CBD and controls. The top enrichment of genes was for JAK ( Janus kinase)–STAT (signal transducer and activator of transcription) signalling. -

Dendritic Cell Biology and Its Role in Tumor Immunotherapy Yingying Wang1,2,3,4† , Ying Xiang2,3†, Victoria W
Wang et al. Journal of Hematology & Oncology (2020) 13:107 https://doi.org/10.1186/s13045-020-00939-6 REVIEW Open Access Dendritic cell biology and its role in tumor immunotherapy Yingying Wang1,2,3,4† , Ying Xiang2,3†, Victoria W. Xin5†, Xian-Wang Wang2,6, Xiao-Chun Peng2,7, Xiao-Qin Liu2,3,8, Dong Wang2,3,NaLi9, Jun-Ting Cheng2,3, Yan-Ning Lyv10, Shu-Zhong Cui1, Zhaowu Ma2,3*, Qing Zhang11,12* and Hong-Wu Xin2,3,13* Abstract As crucial antigen presenting cells, dendritic cells (DCs) play a vital role in tumor immunotherapy. Taking into account the many recent advances in DC biology, we discuss how DCs (1) recognize pathogenic antigens with pattern recognition receptors through specific phagocytosis and through non-specific micropinocytosis, (2) process antigens into small peptides with proper sizes and sequences, and (3) present MHC-peptides to CD4+ and CD8+ T cells to initiate immune responses against invading microbes and aberrant host cells. During anti-tumor immune responses, DC-derived exosomes were discovered to participate in antigen presentation. T cell microvillar dynamics and TCR conformational changes were demonstrated upon DC antigen presentation. Caspase-11-driven hyperactive DCs were recently reported to convert effectors into memory T cells. DCs were also reported to crosstalk with NK cells. Additionally, DCs are the most important sentinel cells for immune surveillance in the tumor microenvironment. Alongside DC biology, we review the latest developments for DC- based tumor immunotherapy in preclinical studies and clinical trials. Personalized DC vaccine-induced T cell immunity, which targets tumor-specific antigens, has been demonstrated to be a promising form of tumor immunotherapy in patients with melanoma. -
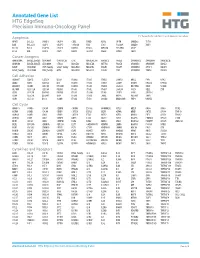
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 -

The Tumor Microenvironment Innately Modulates Cancer Progression Dominique C
Published OnlineFirst July 26, 2019; DOI: 10.1158/0008-5472.CAN-18-3962 Cancer Review Research The Tumor Microenvironment Innately Modulates Cancer Progression Dominique C. Hinshaw1 and Lalita A. Shevde1,2 Abstract Cancer development and progression occurs in concert phoid cells, myeloid-derived suppressor cells, and natural with alterations in the surrounding stroma. Cancer cells can killer cells) as well as adaptive immune cells (T cells and B functionally sculpt their microenvironment through the cells) contribute to tumor progression when present in the secretion of various cytokines, chemokines, and other fac- tumor microenvironment (TME). Cross-talk between cancer tors. This results in a reprogramming of the surrounding cells and the proximal immune cells ultimately results in an cells, enabling them to play a determinative role in tumor environment that fosters tumor growth and metastasis. survival and progression. Immune cells are important con- Understanding the nature of this dialog will allow for stituents of the tumor stroma and critically take part in this improved therapeutics that simultaneously target multiple process. Growing evidence suggests that the innate immune components of the TME, increasing the likelihood of favor- cells (macrophages, neutrophils, dendritic cells, innate lym- able patient outcomes. Introduction edge to current therapies that target dysfunctional cells in the TME. In this review, we summarize the current knowledge on the ability The tumor microenvironment (TME) is complex and contin- of the TME to co-opt innate immune cells for cancer promotion uously evolving. In addition to stromal cells, fibroblasts, and and clinical strategies targeting these innate immune responses in endothelial cells, the TME comprises innate and adaptive immune the context of cancer. -

The Quest for Faithful in Vitro Models of Human Dendritic Cells Types Xin-Long Luo, Marc Dalod
The quest for faithful in vitro models of human dendritic cells types Xin-Long Luo, Marc Dalod To cite this version: Xin-Long Luo, Marc Dalod. The quest for faithful in vitro models of human dendritic cells types. Molecular Immunology, Elsevier, 2020, 123, pp.40-59. 10.1016/j.molimm.2020.04.018. hal-02981716 HAL Id: hal-02981716 https://hal.archives-ouvertes.fr/hal-02981716 Submitted on 30 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. The quest for faithful in vitro models of human dendritic cells types. Luo XL, Dalod M. Mol Immunol. 2020 Jul;123:40‐59. doi: 10.1016/j.molimm.2020.04.018. Epub 2020 May 13. PMID: 32413788. https://www.sciencedirect.com/science/article/abs/pii/S0161589019309174 The quest for faithful in vitro models of human dendritic cells types Xin-Long Luo1 and Marc Dalod1* 1Aix Marseille Univ, CNRS, INSERM, CIML, Centre d'Immunologie de Marseille-Luminy, Marseille, France *Corresponding author at: Centre d’Immunologie de Marseille-Luminy (CIML), Parc scientifique et technnologique de Luminy, case 906, 163 avenue de Luminy, F-13288 Marseille Cedex 09, France. -

Innate Immune Responses to Cryptococcus
Journal of Fungi Review Innate Immune Responses to Cryptococcus Lena J. Heung Infectious Diseases Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA; [email protected]; Tel.: +1-212-639-7980 Received: 24 May 2017; Accepted: 29 June 2017; Published: 2 July 2017 Abstract: Cryptococcus species are encapsulated fungi found in the environment that predominantly cause disease in immunocompromised hosts after inhalation into the lungs. Even with contemporary antifungal regimens, patients with cryptococcosis continue to have high morbidity and mortality rates. The development of more effective therapies may depend on our understanding of the cellular and molecular mechanisms by which the host promotes sterilizing immunity against the fungus. This review will highlight our current knowledge of how Cryptococcus, primarily the species C. neoformans, is sensed by the mammalian host and how subsequent signaling pathways direct the anti-cryptococcal response by effector cells of the innate immune system. Keywords: Cryptococcus; innate immune response; fungal recognition; host-fungus interactions 1. Introduction The encapsulated, yeast-like fungi of the genus Cryptococcus are prevalent throughout the environment worldwide. The most common species that cause disease in humans are Cryptococcus neoformans and Cryptococcus gattii. These pathogens can cause a life-threatening meningoencephalitis after acquisition through the respiratory tract and subsequent dissemination to the central nervous system (CNS). While C. gattii can infect apparently immunocompetent hosts, C. neoformans is more often an opportunistic pathogen, affecting immunocompromised patients including those with HIV/AIDS, cancer and solid organ transplantation [1]. Cryptococcal meningitis has been estimated to affect up to 1 million people worldwide each year [2,3]. -

Signalling Through C-Type Lectin Receptors
REVIEWS Signalling through C-type lectin receptors: shaping immune responses Teunis B. H. Geijtenbeek and Sonja I. Gringhuis Abstract | C‑type lectin receptors (CLRs) expressed by dendritic cells are crucial for tailoring immune responses to pathogens. Following pathogen binding, CLRs trigger distinct signalling pathways that induce the expression of specific cytokines which determine T cell polarization fates. Some CLRs can induce signalling pathways that directly activate nuclear factor‑κB, whereas other CLRs affect signalling by Toll‑like receptors. Dissecting these signalling pathways and their effects on host immune cells is essential to understand the molecular mechanisms involved in the induction of adaptive immune responses. In this Review we describe the role of CLR signalling in regulating adaptive immunity and immunopathogenesis and discuss how this knowledge can be harnessed for the development of innovative vaccination approaches. Dendritic cells (DCs) are located throughout the body Triggering of several PRRs simultaneously can induce T helper (TH) cell A T cell subset that secretes a to capture and internalize invading pathogens, and diverse innate immune responses, which provides the distinct set of cytokines after subsequently process and present antigen on MHC diversity that is required to shape an effective adaptive activation, which occurs class I and class II molecules to CD8+ and CD4+ T cells, immune response. Distinct pathogens express different through the ligation of the T cell respectively1. Antigen presentation by DCs is in itself PAMPs, and the combination of these PAMPs functions receptor with its cognate ligand (peptide–MHC complex), not sufficient to induce effective T cell responses against as a fingerprint that triggers a specific set of PRRs, lead‑ + together with the recognition of pathogens. -

Innate Immune Responses in Cattle Mini Review
Innate Immune Responses in Cattle Mini Review Bovine Innate Immunity This article gives an overview of the bovine innate immune system, examining the roles played by natural killer cells, dendritic cells, and macrophages. Extra attention is given to dendritic cells, including a comparison to human and mouse models. The bovine immune system is made up of two arms: an immunity does not depend on encountering a pathogen; innate (native) response which produces an immediate it is not augmented by repeated exposures to the same reaction and the slower adaptive (acquired) response which pathogen as it has no memory (Rainard and Riollet 2006). occurs 10-14 days post exposure. There are two arms to innate immunity; the sensing arm The innate system functions through a combination of (deals with how the host perceives infection) and the natural defensive barriers - mainly skin, phagocytes and effector arm (deals with how the host combats infection) neutrophils, natural killer cells, cytokines, complement (Beutler 2004). Each arm has a humoral and a cellular and anti-microbial peptides. The sections below will cover component. natural killer cells, macrophages, and dendritic cells making comparisons to human and mouse models. Delineating the innate immune system from the adaptive immune system is very difficult as they share many effector Innate immunity mechanisms (Rainard and Riollet 2006). The innate immune After microbial invasion, sentinel cells, such as system is made up of monocytes, macrophages, dendritic macrophages and dendritic cells, secrete cytokines – cells, natural killer cells, neutrophils and eosinophils. among them interleukin-1 (IL-1) and IL-6, tumour necrosis factor alpha (TNF-α) and high-mobility group protein B1 Natural Killer Cells (HMGB1). -

Ligon Et Al Supplemental Table 3.Xlsx
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Immunother Cancer Supplemental table 3: GSEA (Figure 4B&C, Figure 5A&B) Figure 4B: Immunoregulatory molecules, stimulatory NAME PROBE DESCRIPTION<br>(from dataset) GENE SYMBOL GENE_TITLE RANK IN GENE LIST RANK METRIC SCORE RUNNING ES CORE ENRICHMENT row_0 SELP selectin P [Source:HGNC Symbol;Acc:HGNC:10721] SELP selectin P [Source:HGNC Symbol;Acc:HGNC:10721] 158 1.041440248 0.08885493 Yes row_1 CD40LG CD40 ligand [Source:HGNC Symbol;Acc:HGNC:11935] CD40LG CD40 ligand [Source:HGNC Symbol;Acc:HGNC:11935] 404 0.75878495 0.14717077 Yes row_2 ICOS inducible T cell costimulator [Source:HGNC Symbol;Acc:HGNC:5351] ICOS inducible T cell costimulator [Source:HGNC Symbol;Acc:HGNC:5351] 604 0.649174452 0.1975872 Yes row_3 PRF1 perforin 1 [Source:HGNC Symbol;Acc:HGNC:9360] PRF1 perforin 1 [Source:HGNC Symbol;Acc:HGNC:9360] 660 0.625475645 0.2529252 Yes row_4 CX3CL1 C-X3-C motif chemokine ligand 1 [Source:HGNC Symbol;Acc:HGNC:10647] CX3CL1 C-X3-C motif chemokine ligand 1 [Source:HGNC Symbol;Acc:HGNC:10647] 733 0.599943995 0.30505255 Yes row_5 CXCL9 C-X-C motif chemokine ligand 9 [Source:HGNC Symbol;Acc:HGNC:7098] CXCL9 C-X-C motif chemokine ligand 9 [Source:HGNC Symbol;Acc:HGNC:7098] 739 0.598658085 0.36037394 Yes row_6 IL1A interleukin 1 alpha [Source:HGNC Symbol;Acc:HGNC:5991] IL1A interleukin 1 alpha [Source:HGNC Symbol;Acc:HGNC:5991] 750 0.593991816