Paraneoplastic Syndromes Secondary to Neuroendocrine Tumours
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Drugs for the Treatment of Hypercalcaemia C. R. PATERSON
Postgrad Med J: first published as 10.1136/pgmj.50.581.158 on 1 March 1974. Downloaded from Postgraduate Medical Journal (March 1974) 50, 158-162. Drugs for the treatment of hypercalcaemia C. R. PATERSON M.A., D.M., M.R.C.PATH. Department of Clinical Chemistry, University of Dundee, Dundee Summary able commercially (Phosphate Sandoz). The initial Hypercalcaemia is ordinarily treated by treatment of dose is 1-3 g (of phosphorus) per day according to the underlying disorder. In some cases, as in malignant the size of the patient. The serum calcium should be disease, in vitamin D poisoning and after a failed checked daily at first and the dose adjusted appro- parathyroidectomy, the hypercalcaemia itself needs to priately so that the maintenace dose is the minimum be treated. A large number of methods have been consistent with control of the serum calcium. Oral advocated for this, but phosphate is the drug of choice phosphate therapy is effective in the management of in most patients. This paper outlines its use, mode of hypercalcaemia of malignant disease and myeloma action and side effects and reviews the other methods (Goldsmith and Ingbar, 1966; Goldsmith et al., proposed for the management of hypercalcaemia. 1968; Thalassinos and Joplin, 1968), in hyperpara- thyroidism (Dent, 1962; Goldsmith and Ingbar, 1966; Eisenberg, 1968), and in vitamin D poisoningProtected by copyright. Introduction (Goldsmith and Ingbar, 1966). The management of hypercalcaemia is ordinarily Intravenous phosphate therapy is used in the that of the underlying disorder (Wills, 1971; management of acute hypercalcaemia, especially in Henrard, 1971; Watson, 1972). There are, however, the patient who is vomiting or in coma. -

A Case of Cushing Syndrome with Both Secondary Hypothyroidism and Hypercalcemia Due to Postoperative Adrenal Insufficiency
Endocrine Journal 2004, 51 (1), 105–113 NOTE A Case of Cushing Syndrome with Both Secondary Hypothyroidism and Hypercalcemia Due to Postoperative Adrenal Insufficiency MASAHITO KATAHIRA, TSUTOMU YAMADA* AND MASAHIKO KAWAI* Department of Internal Medicine, Kyoritsu General Hospital, Nagoya 456-8611, Japan *Division of Endocrinology, Department of Internal Medicine, Okazaki City Hospital, Okazaki 444-8553, Japan Abstract. A 48-year-old woman was referred to our hospital because of secondary hypothyroidism. Upon admission a left adrenal tumor was also detected using computed tomography. Laboratory data and adrenal scintigraphy were compatible with Cushing syndrome due to the left adrenocortical adenoma, although she showed no response to the TRH stimulation test. Hypercortisolism resulting in secondary hypothyroidism was diagnosed. After a left adrenalectomy, hydrocortisone administration was begun and the dose was reduced gradually. After discharge on the 23rd postoperative day, she began to suffer from anorexia. ACTH level remained low, and serum cortisol, free thyroxine and TSH levels were within the normal range. Since her condition became worse, she was re-admitted on the 107th postoperative day at which time serum calcium level was high (15.6 mg/dl). Both ACTH response to the CRH stimulation test and TSH response to the TRH stimulation test were restored to almost normal levels, but there was no response of cortisol to CRH stimulation test. We diagnosed that the hypercalcemia was due to adrenal insufficiency. Although the serum calcium level decreased to normal after hydrocortisone was increased (35 mg/day), secondary hypothyroidism recurred. It was suggested that sufficient glucocorticoids suppressed TSH secretion mainly at the pituitary level, which resulted in secondary (corticogenic) hypothyroidism. -

PARANEOPLASTIC SYNDROMES: J Neurol Neurosurg Psychiatry: First Published As 10.1136/Jnnp.2004.040378 on 14 May 2004
PARANEOPLASTIC SYNDROMES: J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.2004.040378 on 14 May 2004. Downloaded from WHEN TO SUSPECT, HOW TO CONFIRM, AND HOW TO MANAGE ii43 J H Rees J Neurol Neurosurg Psychiatry 2004;75(Suppl II):ii43–ii50. doi: 10.1136/jnnp.2004.040378 eurological manifestations of cancer are common, disabling, and often multifactorial (table 1). The concept that malignant disease can cause damage to the nervous system Nabove and beyond that caused by direct or metastatic infiltration is familiar to all clinicians looking after cancer patients. These ‘‘remote effects’’ or paraneoplastic manifestations of cancer include metabolic and endocrine syndromes such as hypercalcaemia, and the syndrome of inappropriate ADH (antidiuretic hormone) secretion. Paraneoplastic neurological disorders (PNDs) are remote effects of systemic malignancies that affect the nervous system. The term PND is reserved for those disorders that are caused by an autoimmune response directed against antigens common to the tumour and nerve cells. PNDs are much less common than direct, metastatic, and treatment related complications of cancer, but are nevertheless important because they cause severe neurological morbidity and mortality and frequently present to the neurologist in a patient without a known malignancy. Because of the relative rarity of PND, neurological dysfunction should only be regarded as paraneoplastic if a particular neoplasm associates with a remote but specific effect on the nervous system more frequently than would be expected by chance. For example, subacute cerebellar ataxia in the setting of ovarian cancer is sufficiently characteristic to be called paraneoplastic cerebellar degeneration, as long as other causes have been ruled out. -

Paraneoplastic Syndromes in Patients with Ovarian Neoplasia
202 Journal of the Royal Society of Medicine Volume 86 April 1993 Paraneoplastic syndromes in patients with ovarian neoplasia C N Hudson MChir FRCS FRCOG1 Marigold Curling MB BS2 Penelope Potsides' D G Lowe MD MRCPath MIBiol' 'The Association of Obstetricians and Gynaecologists, NE Thames Region and 2the Department ofHistopathology, St Bartholomew's Hospital Medical College, London EClA 7BE Keywords: paraneoplastic syndromes; ovarian cancer; prevalence Summary data at presentation of 908 patients with primary The prevalence of several paraneoplastic syndromes epithelial ovarian cancer, collected prospectively in associated with ovarian cancer was determined from the North East Thames Region. a clinicopathological study of 908 patients with primary ovarian malignancy in the North East Thames Data source Region. The diversity and rarity of these manifesta- In the 1970s a data bank for ovarian cancer in the tions are great and the explanation for them is North East Metropolitan Region was set up by difficult. Circumstantial evidence suggests that in the Association of Obstetricians and Gynaecologists some cases an autoimmune phenomenon is the most of the Region in association with the Regional plausible cause. Histopathologists Group. Data was entered either by pathologist or clinician, and as soon as a case Introduction was notified, the clinical data on an agreed proforma Paraneoplastic syndromes are systemic manifestations were obtained from the surgeon concerned - he/ ofcancer that cannot readily be explained by the local she provided details of the mode of presentation, or metastatic effects of a tumour or of hormones investigation, operative staging, and treatment. indigenous to the tissue in which the tumour arises. Histological material was reviewed centrally by The syndromes fall into four broad groups, in which the two of the authors. -
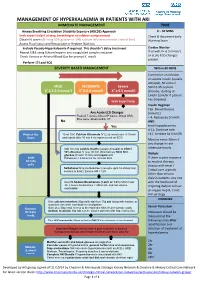
Management of Hyperkalaemia in Patients with Aki Immediate Management Time
MANAGEMENT OF HYPERKALAEMIA IN PATIENTS WITH AKI IMMEDIATE MANAGEMENT TIME ≥ Airway Breathing Circulation Disability Exposure (ABCDE) Approach 0 – 30 MINS Seek expert help if airway, breathing or circulation compromised Check & document Early Obtain IV access (If using 50% glucose or 10% calcium infusions consider central line) Warning Score Assess Fluid status and Resuscitate or Replace fluid loss Exclude Pseudo-Hyperkalaemia if required. This shouldn’t delay treatment Cardiac Monitor Repeat U&E using lithium heparin anti-coagulated sample container If unwell, K+ 6.5 mmol/L + or acute ECG changes Check Venous or Arterial Blood Gas for prompt K result P present Perform 12 Lead ECG SEVERITY BASED MANAGEMENT Within 60 MINS Commence an infusion of soluble insulin (usually actrapid), 50 units in MILD MODERATE Severe 50ml 0.9% sodium + + + K 5.5-5.9 mmol/l K 6-6.4 mmol/l K ≥ 6.5 mmol/l chloride, starting at 1ml/hr (2ml/hr if patient has diabetes) Seek Expert help Insulin Regimen Cap. Blood Glucose Any Acute ECG Changes (mmol/L) Peaked T waves, Absent P waves, Broad QRS, < 4: Reduce by 0.5ml/h Sine wave, Bradycardia, VT No AND Yes treat hypoglycaemia 4-11: Continue rate Protect the 10 ml 10% Calcium Gluconate IV (2.26 mmol) over 5-10 min >11: Increase by 0.5ml/h Heart and repeat after 10 min if no improvement on ECG Monitor every 30min if any change in rate Add 10 units soluble Insulin (usually Actrapid) to 250ml otherwise hourly 10% Glucose IV over 30 min (Alternatively 50ml 50% glucose IV over 15 min) and repeat until Dialysis Shift Potassium < 5.5mmol/L for at least 4hrs. -

Paraneoplastic Hypercalcaemia Associated with TCC of Bladder
Case Study TheScientificWorldJOURNAL (2004) 4, 1069–1070 ISSN 1537-744X; DOI 10.1100/tsw.2004.187 Paraneoplastic Hypercalcaemia Associated with TCC of Bladder A.B. Patel1,*, L. Wilson2, C. Blick2, and P. Meffan2 1Institute of Urology and Nephrology, UCL Medical School, Charles Bell House, 67 Riding House Street, London; 2Department of Urology, East Surrey Hospital, Redhill, Surrey and Sussex NHS Trust, U.K. E-mail: [email protected] Received July 14, 2004; Revised September 28, 2004; Accepted October 5, 2004; Published December 10, 2004 KEYWORDS: TCC, hypercalcaemia,paraneoplastic syndrome DOMAIN: urology Hypercalcaemia associated with transitional carcinoma of the bladder is rare in the absence of metastatic disease. We report a case of paraneoplastic hypercalcaemia associated with a high-grade, organ-confined bladder carcinoma, which resolved upon surgical resection of the tumour. CASE REPORT A 58-year-old woman presented with a 3-week history of weight loss, general malaise, and constipation as well as two episodes of macroscopic haematuria. Laboratory testing on admission to hospital showed marked hypercalcaemia (3.66, normal 2.15–2.60) associated with a PTH level of 1.3 (normal 0.5–5.5) and acute renal failure (BUN 37.5, creatinine 280). Ultrasound examination showed moderate right hydroureteronephrosis associated with a large, lobulated echogenic mass in the bladder. Immediate treatment involved intravenous colloid infusion, intravenous furosemide, and intravenous pamidronate in order to treat the hypercalcaemia. Subsequent transurethral bladder resection revealed grade 3 pT2 transitional cell carcinoma (TCC). Staging computerised tomography and bone scan showed no evidence of local or distant metastatic disease. The patient then underwent radical cystectomy and ileal conduit formation. -

Artefactual Serum Hyperkalaemia and Hypercalcaemia in Essential Thrombocythaemia J Clin Pathol: First Published As 10.1136/Jcp.53.2.105 on 1 February 2000
J Clin Pathol 2000;53:105–109 105 Artefactual serum hyperkalaemia and hypercalcaemia in essential thrombocythaemia J Clin Pathol: first published as 10.1136/jcp.53.2.105 on 1 February 2000. Downloaded from M R Howard, S Ashwell, L R Bond, I Holbrook Abstract the release of potassium ions from platelets Aim—To investigate possible abnormali- which are entrapped within the clot in a serum ties of serum potassium and calcium sample. This phenomenon generally occurs levels in patients with essential thrombo- with platelet counts in excess of 600 × 109/litre, cythaemia and significant thrombocyto- with a roughly predictable increment in serum sis. potassium for every further increase in platelet Methods—24 cases of essential thrombo- count. The potassium level is normalised if the cythaemia with significant thrombocyto- estimation is made using plasma rather than sis (platelet count > 700 × 109/litre) had serum.45 serum potassium and calcium estimations Abnormalities of other ions have not been performed at the time of maximum well described in essential thrombocythaemia. thrombocytosis before treatment, and at Despite the presence of calcium in platelet- the time of low platelet count after dense granules and its secretion from platelets treatment with cytoreductive drugs. Se- during activation, there has been no systematic lected patients were further investigated study of serum calcium levels in patients with with plasma sampling and estimation of essential thrombocythaemia and significantly ionised calcium and parathyroid hor- increased platelet counts. There has been a mone. single case report of serum hypercalcaemia Results—At the time of maximum throm- associated with essential thrombocythaemia. In bocytosis six patients had serum hyperka- this case the hypercalcaemia rapidly resolved laemia (> 5.5 mmol/litre) and five had following reduction of the platelet count.6 We serum hypercalcaemia (> 2.6 mmol/litre). -

Thymoma: an Update DR CESAR MORAN
TUEDAY SURGICAL PATHOLOGY Thymoma: an update DR CESAR MORAN TUEDAY SURGICAL PATHOLOGY Glial and Glial-Neuronal Tumors DR ARIE PERRY GLIAL AND GLIO- NEURONAL TUMORS Arie Perry, M.D. DISCLOSURES (Arie Perry, MD) • I have no financial relationships to disclose. - and - • I will not discuss off label use or investigational use in my presentation School of Medicine NEUROPATHOLOGY AND REAL ESTATE • Location • Location • Location • Patient Age • Neuroimaging School of Medicine GLIOMAS • Astrocytomas (A) • Oligodendrogliomas (O) • [Mixed oligoastrocytomas (MOA)] • Ependymomas • Diffuse glioma = A, O, or MOA School of Medicine GLIOMA GRADING: WHO 2016 • Grade I = Benign • Grade II = Low-grade • Grade III = “Anaplastic” • Grade IV = High-grade malignant, e.g. “GBM” School of Medicine Distribution of Primary Malignant Brain and Other CNS Tumors by CBTRUS Histology Groupings and Histology and Behavior (N = 121,277) CBTRUS Statistical Report: NPCR and SEER, 2011–2015. Gliomas account for 26% of all primary CNS tumors, but 81% of all malignant CNS tumors Neuro Oncol. 2018;20(suppl_4):iv1-iv86. doi:10.1093/neuonc/noy131 © The Author(s) 2018. Published by Oxford University Press on behalf of the Society for Neuro-Oncology. ASTROCYTOMAS • Diffuse (75%) • Circumscribed / Favorable (25%) – Fibrillary – Pilocytic – Gemistocytic – PXA – Giant Cell – SEGA – Small Cell – DIA – Granular Cell – Epithelioid School of Medicine ASTROCYTOMA, IDH-mutant, WHO GRADE II • Age 30-40 • Insidious / Slow growing • Non-enhancing • Often progress to grades III or IV • Survival -

Multiple Myeloma Associated With
ISSN: 2469-5696 Islam and Kotoucek. Int J Blood Res Disord 2018, 5:029 DOI: 10.23937/2469-5696/1410029 International Journal of Volume 5 | Issue 1 Open Access Blood Research and Disorders RESEARCH ARTICLE Multiple Myeloma Associated with Dermatomyositis: A Short Report and Mini-Review Md Serajul Islam1,2* and Pavel Kotoucek2 Check for 1Department of Haematology, Guys’ & St Thomas’ Hospital, London, UK updates 2Department of Haematology, Broomfield Hospital, Chelmsford, UK *Corresponding author: Md Serajul Islam, Department of Haematology, Guys’ & St Thomas’ Hospital, London, UK; De- partment of Haematology, Broomfield Hospital, Chelmsford, UK, Tel: +44-7769580452, Fax: +44-1245-51-6669, E-mail: [email protected] amyloid deposits in various tissues [3]. The American Abstract Cancer Society has estimated that 30,770 new MM Multiple myeloma (MM) is characterized by the neoplastic proliferation of plasma cell clones that produce monoclo- cases will occur in the United States in 2018, with an nal immunoglobulin. Dermatomyositis (DM), and to a lesser estimated 12,770 deaths [4]. Idiopathic inflammatory extent polymyositis (PM), carry a higher risk of cancer than myopathies (IIM), a heterogeneous group of disorders that of the general population as demonstrated by several characterised by weakness and inflammation of skele- studies with the prevalence being 32% and 15% for DM and PM respectively. The mechanism underlying the association tal muscle, are often associated with malignancies [5]. between idiopathic inflammatory myopathies (IIM) and ma- Polymyositis (PM) and dermatomyositis (DM) are clas- lignancies remains unclear. Both conditions predominantly sified as IIM, with these conditions predominantly af- affect adults. About 15% of DM is refractory to conventional fecting adults [6]. -

Hypercalcemia
Hypercalcemia Dr. Andrés Pérez Riera Causes: Hypercalcemia is divided into PTH-mediated hypercalcemia (primary hyperparathyroidism) and non–PTH-mediated hypercalcemia. 1) PTH-mediated hypercalcemia is related to increased calcium absorption from the intestine. 2) Non–PTH-mediated hypercalcemia includes the following: (2.1) Hypercalcemia associated with malignancy: Unlike PTH-mediated hypercalcemia, the elevation of calcium that results from malignancy generally worsens until therapy is provided. Hypercalcemia caused by malignancy is the result of increased osteoclastic activity within the bone. This results from one or both of the mechanisms that follow: (2.1-a) Extensive localized bone destruction may result from osteolytic metastasis of solid tumors. Evidence indicates that many malignant cells may release local osteoclastic activating factors. (2.1-b) Increased calcium levels resulting from malignancy caused by a PTH- related protein is a second mechanism. This protein is a humeral factor that acts on the skeleton to increase bone reabsorption; it acts on the kidney to decrease excretion of calcium. The gene that produces this protein is present in many malignant tissues. (2.2) Granulomatous disorders: High levels of calcitriol may be found in patients with sarcoidosis and other granulomatous diseases. In these disorders, the increased level of calcitriol results from production within the macrophages, which constitute a large portion of some granulomas. (2.3) Iatrogenic: In some cases, elevation of calcium is a known adverse effect of appropriate dosage. In other cases, large ingestions must be taken to induce the increase in calcium levels. Obtain a complete review of current medications for patients presenting with hypercalcemia. Record any vitamin use. -

Hypercalcaemia As an Indication of Adrenal Insufficiency in A
HYPERCALCAEMIA AS AN INDICATION OF ADRENAL INSUFFICIENCY IN A PATIENT WITH AUTOIMMUNE POLYENDOCRINOPATHY-CANDIDIASIS- ECTODERMAL DYSTROPHY (APECED) Dikaiakou E.¹, Vlachopapadopoulou E.¹, Anagnostou E.¹, Panagiotopoulos I.¹, Fotinou A.², Michalacos S.¹ 1.Dept. of Endocrinology-Growth and Development, Children's Hospital P. & A. Kyriakou, Athens, Greece 2. Biochemistry Dept.-Hormones Laboratory, Children's Hospital P. & A. Kyriakou, Athens, Greece Introduction Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), is a rare inherited disease of childhood, caused by the mutation of the AIRE gene on chromosome 21. It is characterized by 3 main diseases: chronic mucocutaneous candidiasis (CMC), chronic hypoparathyroidism (HP) and Addison’s disease (AD), and can be associated with other autoimmune diseases and/or manifestations of ectodermal dystrophy. Case presentation AIRE gene mutations (a) and functional domains of corresponding proteins (b) An 8 year old girl, who was known to have hypoparathyroidism and she was treated with calcium and calcitriol, presented at the endocrinology department, complaining of fatigue and abdominal pain for five days, without vomiting, nor fever. Physical examination On physical examination she was pale with poor skin turgor, low normal blood pressure and mildly tachycardic. Laboratory findings The initial blood investigation revealed: 1.hypercalcaemia 11.6 mg/dl 2.hyponatremia 125 m Eq/l Clinical features of AIRE deficiency/APECED 3.hyperkalemia 5.8 m Eq/l which suggested the possibility of adrenal failure. The classic triad is Further investigation mucocutaneous candidiasis, Further laboratory investigation demonstrated: hypoparathyroidism, and 1.elevated ACTH levels (3465 pg/ml) 2.decreased cortisol production (5.84 μg/dl) adrenal failure 3.anti-adrenal antibodies were present. -

Hypocalcaemia in Vit D Deficiency Or If PTH Suppressed
StR Teaching in GIM 19th September 2017 Hypercalcaemia: Diagnosis, acute management and cases Afroze Abbas BSc PhD FRCP Consultant Endocrinologist [email protected] 2017 1 What should a GIM StR know about calcium disorders? HYPERCALCAEMIA • Diagnostic approach • Symptoms and signs • Causes • Mechanisms • Acute management • Cases • Questions! [email protected] 2017 2 Case 1: Presentation • 23 year old woman • Readmitted 2 days later • Presents to A&E with extremely dehydrated nausea, vomiting and and unwell. abdominal pain • Adjusted calcium: • Pregnancy test negative 4.20mmol/L (2.20- 2.60mmol/L) • Venous gas: normal pH, HCO3- slightly low, • U&E consistent with calcium 2.3mmol/L dehydration • Diagnosed with gastroenteritis and discharged. [email protected] 2017 3 Hypercalcaemia • Common • 90% cases: • Malignancy • Primary Hyperparathyroidism • Entry of calcium into blood > urinary calcium excretion + calcium bone deposition • Mechanisms: • Accelerated bone resorption • Excessive GI absorption • Decreased renal calcium excretion [email protected] 2017 4 Calcium Homeostasis • Tightly regulated • Muscle function • Intracellular signalling • Neuronal function • Coagulation • 45% bound to proteins (albumin) • 15% bound to anions (phosphate and citrate) • 40% free/ionised calcium – metabolically active [email protected] 2017 5 Calcium Homeostasis • Calcium range 2.20-2.60mmol/L (variable) • ↓ Alb 1g/dL = ↓Calcium 0.2mmol/L • Volume overload, illness, nephrotic syndrome, malnutrition • Adjusted-calcium better measure in hypo/