Cryptic and Extensive Hybridization Between Ancient Lineages of American Crows
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bird Studies Overview
Chapter 7. Bird Studies Overview teigerwald Lake National Wildlife Refuge provides a variety of habitats for many species of birds. Thousands of breeding birds rely on the resources of the refuge to Srest, eat, and raise their young. In addition, the refuge supports wetlands that are vital to the survival of migratory birds. The activities that follow offer an excellent opportunity for students to learn about and to observe the different species of birds — their behaviors and adaptations to the habitats on the refuge. Background The actively managed refuge wetlands and grasslands, when combined with the natural floodplain vegetative communities, provide habitat that supports over 200 species of birds. Hundreds of thousands of birds migrate along the lower Columbia River every year. The refuge hosts thousands of migratory birds that fly thousands of miles from their breeding grounds in Arctic Canada and Alaska to their wintering grounds in Baja California or South America, a route known as the Pacific Flyway. The few remaining areas of wetland habitat along the lower Columbia River are vital to the flyway. Some birds spend their winter on refuge wetlands, returning north to nest; some nest here but migrate to milder climates in the south for the winter; and some do not migrate at all but remain in the area as permanent residents. Several of the songbirds found in the summer spend our winters in Central and South America, migrating thousands of miles annually between their summer and winter habitats. Birds using the refuge are specifically adapted to the type of food they eat and the type of habitat they occupy (open water, freshwater wetland, field, riparian woodland, or upland woodland). -

Crows and Ravens Wildlife Notes
12. Crows & Ravens Crows and ravens belong to the large family Corvidae, along with more than 200 other species including jays, nutcrackers and magpies. These less-than-melodious birds, you may be surprised to learn, are classified as songbirds. raven American Crow insects, grain, fruit, the eggs and young of other birds, Crows are some of the most conspicuous and best known organic garbage and just about anything that they can find of all birds. They are intelligent, wary and adapt well to or overpower. Crows also feed on the carcasses of winter – human activity. As with most other wildlife species, crows and road-killed animals. are considered to have “good” points and “bad” ones— value judgements made strictly by humans. They are found Crows have extremely keen senses of sight and hearing. in all 50 states and parts of Canada and Mexico. They are wary and usually post sentries while they feed. Sentry birds watch for danger, ready to alert the feeding birds with a sharp alarm caw. Once aloft, crows fly at 25 Biology to 30 mph. If a strong tail wind is present, they can hit 60 Also known as the common crow, an adult American mph. These skillful fliers have a large repertoire of moves crow weighs about 20 ounces. Its body length is 15 to 18 designed to throw off airborne predators. inches and its wings span up to three feet. Both males Crows are relatively gregarious. Throughout most of the and females are black from their beaks to the tips of their year, they flock in groups ranging from family units to tails. -

Predator and Competitor Management Plan for Monomoy National Wildlife Refuge
Appendix J /USFWS Malcolm Grant 2011 Fencing exclosure to protect shorebirds from predators Predator and Competitor Management Plan for Monomoy National Wildlife Refuge Background and Introduction Background and Introduction Throughout North America, the presence of a single mammalian predator (e.g., coyote, skunk, and raccoon) or avian predator (e.g., great horned owl, black-crowned night-heron) at a nesting site can result in adult bird mortality, decrease or prevent reproductive success of nesting birds, or cause birds to abandon a nesting site entirely (Butchko and Small 1992, Kress and Hall 2004, Hall and Kress 2008, Nisbet and Welton 1984, USDA 2011). Depredation events and competition with other species for nesting space in one year can also limit the distribution and abundance of breeding birds in following years (USDA 2011, Nisbet 1975). Predator and competitor management on Monomoy refuge is essential to promoting and protecting rare and endangered beach nesting birds at this site, and has been incorporated into annual management plans for several decades. In 2000, the Service extended the Monomoy National Wildlife Refuge Nesting Season Operating Procedure, Monitoring Protocols, and Competitor/Predator Management Plan, 1998-2000, which was expiring, with the intent to revise and update the plan as part of the CCP process. This appendix fulfills that intent. As presented in chapter 3, all proposed alternatives include an active and adaptive predator and competitor management program, but our preferred alternative is most inclusive and will provide the greatest level of protection and benefit for all species of conservation concern. The option to discontinue the management program was considered but eliminated due to the affirmative responsibility the Service has to protect federally listed threatened and endangered species and migratory birds. -

The Vocal Behavior of the American Crow, Corvus Brachyrhynchos
THE VOCAL BEHAVIOR OF THE AMERICAN CROW, CORVUS BRACHYRHYNCHOS THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Sciences in the Graduate School of The Ohio State University By Robin Tarter, B.S. ***** The Ohio State University 2008 Masters Examination Committee Approved by Dr. Douglas Nelson, Advisor Dr. Mitch Masters _________________________________ Dr. Jill Soha Advisor Evolution, Ecology and Organismal Biology Graduate Program ABSTRACT The objective of this study was to provide an overview of the vocal behavior of the American crow, Corvus brachyrhynchos, and to thereby address questions about the evolutionary significance of crow behavior. I recorded the calls of 71 birds of known sex and age in a family context. Sorting calls by their acoustic characteristics and behavioral contexts, I identified and hypothesized functions for 7 adult and 2 juvenile call types, and in several cases found preferential use of a call type by birds of a particular sex or breeding status. My findings enrich our understanding of crow social behavior. I found that helpers and breeders played different roles in foraging and in protecting family territories from other crows and from predators. My findings may also be useful for human management of crow populations, particularly dispersal attempts using playbacks of crows’ own vocalizations. ii ACKNOWLEDGEMENTS I would like to thank Dr. Kevin McGowan of Cornell, Dr. Anne Clark of Binghamton University, and Binghamton graduate student Rebecca Heiss for allowing me to work with their study animals. McGowan, Clark and Heiss shared their data with me, along with huge amounts of information and insight about crow behavior. -

OWLS of OHIO C D G U I D E B O O K DIVISION of WILDLIFE Introduction O W L S O F O H I O
OWLS OF OHIO c d g u i d e b o o k DIVISION OF WILDLIFE Introduction O W L S O F O H I O Owls have longowls evoked curiosity in In the winter of of 2002, a snowy ohio owl and stygian owl are known from one people, due to their secretive and often frequented an area near Wilmington and two Texas records, respectively. nocturnal habits, fierce predatory in Clinton County, and became quite Another, the Oriental scops-owl, is behavior, and interesting appearance. a celebrity. She was visited by scores of known from two Alaska records). On Many people might be surprised by people – many whom had never seen a global scale, there are 27 genera of how common owls are; it just takes a one of these Arctic visitors – and was owls in two families, comprising a total bit of knowledge and searching to find featured in many newspapers and TV of 215 species. them. The effort is worthwhile, as news shows. A massive invasion of In Ohio and abroad, there is great owls are among our most fascinating northern owls – boreal, great gray, and variation among owls. The largest birds, both to watch and to hear. Owls Northern hawk owl – into Minnesota species in the world is the great gray are also among our most charismatic during the winter of 2004-05 became owl of North America. It is nearly three birds, and reading about species with a major source of ecotourism for the feet long with a wingspan of almost 4 names like fearful owl, barking owl, North Star State. -

U.S. Fish & Wildlife Service Merritt Island CHECKLIST
SP SU FA WN STATUS SP SU FA WN STATUS SP SU FA WN STATUS SP SU FA WN STATUS SP SU FA WN STATUS U.S. Fish & Wildlife Service Gulls, Terns, and Skimmers Nightjars (Goatsuckers) Larks Nashville Warbler R R Dark-eyed Junco 1977 Black-legged Kittiwake R R Common Nighthawk * C C U Horned Lark 1994 Northern Parula * C U C R Lapland Longspur 1988 Merritt Island Sabine's Gull 2009 Chuck-will's-widow * C C O Yellow Warbler U U R Snow Bunting 1999 National Wildlife Refuge Swallows and Martins Bonaparte's Gull 0 U U Whip-poor-will R R Chestnut-sided Warbler O 0 Titusville, Florida Purple Martin * U U R Black-headed Gull 2008 Magnolia Warbler 0 0 Cardinals, Grosbeaks, Buntings, and Allies Swifts Tree Swallow C R C C Laughing Gull* C C C C Cape May Warbler U O Summer Tanager OOO Chimney Swift U U U Northern Rough-winged Swallow U R U Franklin's Gull 2003 Black-throated Blue Warbler U U Scarlet Tanager O O Bank Swallow O O Northern Cardinal * C C C C Ring-billed Gull C R C C Hummingbirds Yellow-rumped Warbler C C C Herring Gull U R U U Ruby-throated Hummingbird U U U R Cliff Swallow O O Black-throated Green Warbler O OO Rose-breasted Grosbeak O O Lesser Black-backed Gull U U U Barn Swallow C 0 C Townsend's Warbler 2002 Blue Grosbeak * OOO Kingfishers Indigo Bunting* U U U Glaucous Gull 1991 Chickadees and Titmice Blackbumian Warbler O O Great Black-backed Gull U 0 U U Belted Kingfisher* C R C C Yellow-throated Warbler U U U Painted Bunting * U O U U Carolina Chickadee 1976 Dickcissel R R R Brown Noddy R R Woodpeckers and Allies Tufted Titmouse * U U -

Hellmayr's 'Catalogue of Birds of the Americas'
Vol.1935 LII] J RecentLiterature. 105 RECENT LITERATURE. Hellmayr's 'Catalogue of Birds of the Americas.'--The long awaitedseventh part of this notable work• has appeared, covering the families Corvidae to Sylviidae in almostthe orderof the A. O. U. 'Check-List,'with the neotropicalfamily Zeledo- niidae betweenthe Turdidae and Sylviidae. The volume follows exactly the style of its predecessors,with the same abundanceof foot notes discussingcharacters and relationshipsof many of the forms, and will, we are sure, prove of the sameimpor- tance to systematic studentsof the avifauna of the Americas. It brings the subject up to July 1, 1932. It is impossiblein the space at our disposalto discussall of the innovations in systematic arrangement and in nomenclaturewhich are presentedby Dr. Hel]mayr in these pages and we shall have •o be content, for the most part, with a comparison of his findingsin regard to North American Birds with those of the A. O. U. reittee, as expressedin the fourth edition of the 'Check-List.' The author is very conservativein his treatment of genera six of those recognized in the 'Check-List' beingmerged with others--Penthestesand Baeolophuswith Parus; Telmatodyteswith Cistothorus;Nahnus with Troglodyles;Arceuthornis with Turdus; Corthyliowith Regulus--going back in every case but one to the treatment of the first edition of the 'Check-List,' in 1886, so doesthe pendulumswing from one ex- treme to the other and back again! Generic division is largely a matter of personal opinion and while there is much to be said in favor of someof Dr. Hellmayr's actions it seemsa pity that two well-markedgroups like the Long- and Short-billed Marsh Wrens should have to be merged simply becauseof the discovery of a more or less intermediate speciesin South America. -

American Crow Corvus Brachyrhynchos
American crow Corvus brachyrhynchos Kingdom: Animalia FEATURES Phylum: Chordata The American crow is a large bird (17 to 21 inches) Class: Aves with a large, strong bill. Its nostrils are covered by Order: Passeriformes bristles. Both the male and the female are entirely black in color. Family: Corvidae ILLINOIS STATUS BEHAVIORS common, native The American crow is a common, statewide, permanent resident of Illinois. Some crows do © U.S. Army Corps of Engineers migrate, and those that migrate start spring migration in February or March. Nesting season occurs during the period March through May with one brood raised per year. The nest is built of sticks, bark and vines and lined with bark, mosses, grasses, feathers and other materials. It is placed in the crotch of a tree or near the tree trunk on a horizontal branch, from 10 to 70 feet above the ground. Both the male and the female construct the nest in a process that takes nearly two weeks. The female lays two to seven green-blue to pale-blue eggs that are marked with darker colors. The incubation period lasts for 18 days, and both male and female share incubation duties. Fall migration includes mainly crows moving into Illinois from more northerly states and northern Illinois crows moving adult into central Illinois. The American crow makes a “caw” noise. It eats corn, sumac berries, poison ivy ILLINOIS RANGE berries, insects, dead animals, eggs and nestlings of other birds and most anything edible. It lives in open or semi-open areas, woodland edges, woodlands, shores, river groves and farm fields. -

A Global Assessment of the Conservation Status of the Black Oystercatcher Haematopus Bachmani
A global assessment of the conservation status of the Black Oystercatcher Haematopus bachmani David F. Tessle r1, James A. Johnso n2, Brad A. Andres 3, Sue Thoma s4 & Richard B. Lancto t2 1Alaska Department of Fish and Game, Division of Wildlife Conservation, Wildlife Diversity Program, 333 Raspberry Road, Anchorage, Alaska 99518 USA. [email protected] 2United States Fish and Wildlife Service, Division of Migratory Bird Management, 1011 East Tudor Road, MS 201, Anchorage, Alaska 99503 USA 3United States Fish and Wildlife Service, Division of Migratory Bird Management, 755 Parfet Street, Suite 235, Lakewood, Colorado 80215 USA 4United States Fish and Wildlife Service, Washington Maritime National Wildlife Refuge Complex, 715 Holgerson Road, Sequim, Washington 98382 USA Tessler, D.F., J.A. Johnson, B.A. Andres, S. Thomas, & R.B. Lanctot. 2014. A global assessment of the conser - vation status of the Black Oystercatcher Haematopus bachmani . International Wader Studies 20: 83 –96. The Black Oystercatcher Haematopus bachmani , a monotypic species, is one of the less studied members of the genus. The global population of roughly 10,000 individuals is scattered unevenly along the North American Pacific Ocean coast from the Aleutian Islands to Baja California, with the vast majority (about 80%) in Alaska and British Columbia. Favouring rocky shorelines in areas of high tidal variation, they forage exclusively on intertidal macroinvertebrates (e.g. limpets and mussels). Because they are completely dependent on marine shorelines, the Black Oystercatcher is considered a sensitive indicator of the health of the rocky intertidal community. Breeding oystercatchers are highly territorial, and nesting densities are generally low; however, during the winter months they tend to aggregate in groups of tens to hundreds. -

Brettsmithersthesis2003.Pdf
Copyright © 2003 Brett Smithers i ACKNOWLEDGMENTS This study would not have been possible without the efforts and contributions of many organizations and individuals. My appreciation is extended to Dr. Clint Boal, my major advisor, for helpful and timely editing, guidance and for serving as my mentor. Thanks are extended to Dr. David Andersen, my co-advisor, for his scholarly and insightful comments and advice. Funding for this project was provided by the Chippewa National Forest, The National Council for Air and Steam Improvement, Minnesota Department of Natural Resources, Potlatch Corporation, Superior National Forest, and U.S. Fish and Wildlife Service. I owe a great deal to those individuals who assisted with nest inventories, trapping, equipment installation, and changing tapes and batteries at monitored nests: Aimee Roberson, Lisa Smithers, Amanda Wester, Wayne Steffans, Frank Nicoletti, Cameron Trembath, Jeremy Ridelbauer, and Ann Bellman. Matt Solensky assisted with hawk trapping, and Steve Day of Airways Aviation provided air service for relocating telemetered goshawks during the 2001 field season. Personnel from the many cooperating agencies and organizations provided assistance and logistical support during this project. They included, in no particular order, Jim Gallagher, John Casson, Jeff Hines, Mike Houser, Rich Baker, Al Williamson, Ben Ohlander, Steve Mortensen, Carol Mortensen, Robin Vora, Maya Hamady, Lissa Grover, Ed Lindquist, and Wayne Russ. ii Video equipment for the 2000 and 2001 field seasons was provided by Alaska Department of Fish & Game, and the Wisconsin Department of Natural Resources. The Minnesota Department of Natural Resources provided office space, computer, and an all- terrain vehicle during the 2002 field season. -

Prey of Breeding Northern Goshawks in Washington
j. RaptorRes. 32(4):297-305 ¸ 1998 The Raptor ResearchFoundation, Inc. PREY OF BREEDING NORTHERN GOSHAWKS IN WASHINGTON JAMESW. WATSON,DAVID W. HAYSAND SEAN P. FINN1 WashingtonDepartment of Fish and Wildlife,600 CapitolWay N., Olympia,WA 98501-1091 U.S.A. PAUL MEEHAN-MARTIN 2 ResearchExperiment Station, 3625 93rd Ave. S.W., Olympia,WA 98502 U.S.A. ABSTRACT.--Weidentified 936 prey from food remains and pellets collected at 82 Northern Goshawk (Accipitergentilis) nest sitesin Washingtonfrom 1986-96. Mammals and birds constitutedhalf of the prey by frequencyand biomassthroughout Washington, although birds were more prevalent(P = 0.050) in the diet of goshawksnesting in the Olympicand Cascademountains of westernWashington (53%), than in the Cascadesof easternWashington (47%). Douglas'squirrels (Tamiasciurus douglasii), grouse (Dendragapusobscurus and Bonasaumbellus), and snowshoehares (Lepusamericanus) were jointly the most frequentlyrepresented prey on the westside (41%) and eastside (54%). Grouseand snowshoehares accountedfor the overwhelmingmajority of prey biomassin theserespective areas (76% and 80%). Relativeto other Northern Goshawkpopulations, goshawks in Washingtonappeared to prey on species from a similar number of genera,but they had a smallerfood-niche breadth and they took larger-sized birds primarily due to their high consumptionof grouse.Northern Goshawksin westernWashington took prey in more equal numbersthan thoseon the eastside. Potential bias from examinationof prey remainswhen comparedto pelletsreinforced the need for inclusionof -
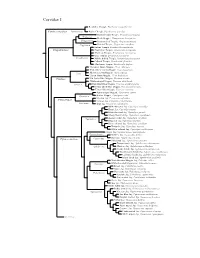
Corvidae Species Tree
Corvidae I Red-billed Chough, Pyrrhocorax pyrrhocorax Pyrrhocoracinae =Pyrrhocorax Alpine Chough, Pyrrhocorax graculus Ratchet-tailed Treepie, Temnurus temnurus Temnurus Black Magpie, Platysmurus leucopterus Platysmurus Racket-tailed Treepie, Crypsirina temia Crypsirina Hooded Treepie, Crypsirina cucullata Rufous Treepie, Dendrocitta vagabunda Crypsirininae ?Sumatran Treepie, Dendrocitta occipitalis ?Bornean Treepie, Dendrocitta cinerascens Gray Treepie, Dendrocitta formosae Dendrocitta ?White-bellied Treepie, Dendrocitta leucogastra Collared Treepie, Dendrocitta frontalis ?Andaman Treepie, Dendrocitta bayleii ?Common Green-Magpie, Cissa chinensis ?Indochinese Green-Magpie, Cissa hypoleuca Cissa ?Bornean Green-Magpie, Cissa jefferyi ?Javan Green-Magpie, Cissa thalassina Cissinae ?Sri Lanka Blue-Magpie, Urocissa ornata ?White-winged Magpie, Urocissa whiteheadi Urocissa Red-billed Blue-Magpie, Urocissa erythroryncha Yellow-billed Blue-Magpie, Urocissa flavirostris Taiwan Blue-Magpie, Urocissa caerulea Azure-winged Magpie, Cyanopica cyanus Cyanopica Iberian Magpie, Cyanopica cooki Siberian Jay, Perisoreus infaustus Perisoreinae Sichuan Jay, Perisoreus internigrans Perisoreus Gray Jay, Perisoreus canadensis White-throated Jay, Cyanolyca mirabilis Dwarf Jay, Cyanolyca nanus Black-throated Jay, Cyanolyca pumilo Silvery-throated Jay, Cyanolyca argentigula Cyanolyca Azure-hooded Jay, Cyanolyca cucullata Beautiful Jay, Cyanolyca pulchra Black-collared Jay, Cyanolyca armillata Turquoise Jay, Cyanolyca turcosa White-collared Jay, Cyanolyca viridicyanus