Arthropod Genomics Symposium (Ags) Thank You to Our Sponsors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Arthropod IGF, Relaxin and Gonadulin, Putative Orthologs of Drosophila
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.11.088476; this version posted June 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Arthropod IGF, Relaxin and Gonadulin, putative 2 orthologs of Drosophila insulin-like peptides 6, 7 and 3 8, likely originated from an ancient gene triplication 4 5 6 Jan A. Veenstra1, 7 8 1 INCIA UMR 5287 CNRS, University of Bordeaux, Bordeaux, Pessac, France 9 10 Corresponding Author: 11 Jan A. Veenstra1 12 INCIA UMR 5287 CNRS, Université de Bordeaux, allée Geoffroy St Hillaire, CS 50023, 33 615 13 Pessac Cedex, France 14 Email address: [email protected] 15 16 Abstract 17 Background. Insects have several genes coding for insulin-like peptides and they have been 18 particularly well studied in Drosophila. Some of these hormones function as growth hormones 19 and are produced by the fat body and the brain. These act through a typical insulin receptor 20 tyrosine kinase. Two other Drosophila insulin-like hormones are either known or suspected to act 21 through a G-protein coupled receptor. Although insulin-related peptides are known from other 22 insect species, Drosophila insulin-like peptide 8, one that uses a G-protein coupled receptor, has 23 so far only been identified from Drosophila and other flies. However, its receptor is widespread 24 within arthropods and hence it should have orthologs. Such putative orthologs were recently 25 identified in decapods and have been called gonadulins. -

Historical, Landscape and Resource Influences on the Coccinellid Community in Missouri
HISTORICAL, LANDSCAPE AND RESOURCE INFLUENCES ON THE COCCINELLID COMMUNITY IN MISSOURI _______________________________________ A Dissertation presented to the Faculty of the Graduate School at the University of Missouri-Columbia _______________________________________________________ In Partial Fulfillment of the requirements for the Degree Doctor of Philosophy _____________________________________________________ by LAUREN M. DIEPENBROCK Dr. Deborah Finke, Dissertation Supervisor MAY 2014 The undersigned, appointed by the Dean of the Graduate School, have examined the dissertation entitled: HISTORICAL, LANDSCAPE AND RESOURCE INFLUENCES ON THE COCCINELLID COMMUNITY IN MISSOURI Presented by Lauren M. Diepenbrock a candidate for the degree of Doctor of Philosophy and hereby certify that in their opinion is worthy of acceptance ________________________________________________ Dr. Deborah Finke, Dissertation Supervisor, Division of Plant Sciences ________________________________________________ Dr. Richard Houseman, Division of Plant Sciences ________________________________________________ Dr. Bruce Barrett, Division of Plant Sciences ________________________________________________ Dr. John Faaborg, Division of Biological Sciences ACKNOWLEDGEMENTS I would like to thank my Ph. D. advisor, Dr. Deborah Finke for the opportunity to pursue a doctoral degree in insect ecology and for her guidance and support throughout my time at the University of Missouri. I would also like to thank my graduate committee, Drs. Houseman, Barrett and Faaborg for their helpful advice during this academic journey. In addition to my graduate committee, I am grateful for the advice and opportunities that were made available to me by Dr. Rose-Marie Muzika, who introduced me to the Conservation Biology certificate program and all of the great researchers across the university who share my interests in biodiversity conservation. I will always be grateful to Dr. Jeanne Mihail for introducing me to Dr. -

(Coleoptera: Coccinellidae) of the Atlantic Maritime Ecozone
Chapter 21 Ladybird beetles (Coleoptera: Coccinellidae) of the Atlantic Maritime Ecozone Christopher G. Majka and David B. McCorquodale Abstract: The Atlantic Maritime Ecozone Coccinellidae (lady beetles) are a conspicuous component of the region’s beetle fauna. Fifty-one species have been recorded, six of which are introduced. In addition, there are records of intentional and inadvertent introductions of 10 species that have not persisted. In this treatment, we discuss the general biology of the Coccinellidae, the fauna of the Atlantic Maritime Ecozone, and the history of collecting efforts for the group. The distri- bution of species (in varying degrees of detail) is examined with a consideration of the differences between provinces. Attention is paid to the zoogeographic factors that influence distribution, including the role of geography, latitude, and the isolation of island faunas. The ecology, distribution, and systematics of individual species are discussed with a consider- ation of the role of both the geological history in shaping the distribution of native species, and human history and agency in the distribution of introduced species. We also consider the impact of introduced species on native faunas and examine the composition of the fauna in terms of global and continental zoogeographic patterns. Résumé : Les coccinellidés (coccinelles) de l’écozone maritime de l’Atlantique sont une composante remarquable de la faune de coléoptères de la région. Cinquante-et-une espèces ont été répertoriées, parmi lesquelles six ont été introduites. Il existe en outre des témoignages d’introductions intentionnelles et non intentionnelles de dix espèces qui n’ont pas per- sisté. Dans ce document, nous abordons la biologie des coccinellidés en général, la faune qui occupe l’écozone maritime de l’Atlantique en particulier, et l’historique des efforts de collecte pour ce groupe. -
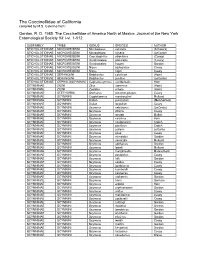
The Coccinellidae of California Compiled by M.S
The Coccinellidae of California compiled by M.S. Caterino from: Gordon, R. D. 1985. The Coccinellidae of America North of Mexico. Journal of the New York Entomological Society 93: i-vi, 1-912. SUBFAMILY TRIBE GENUS SPECIES AUTHOR STICHOLOTIDINAE MICROWEISEINI Microweisea suturalis (Schwarz) STICHOLOTIDINAE MICROWEISEINI Microweisea misella (LeConte) STICHOLOTIDINAE MICROWEISEINI Coccidophilus atronitens (Casey) STICHOLOTIDINAE MICROWEISEINI Gnathoweiea planiceps (Casey) STICHOLOTIDINAE MICROWEISEINI Gnathoweiea hageni Gordon STICHOLOTIDINAE MICROWEISEINI Nipus biplagiatus Casey STICHOLOTIDINAE MICROWEISEINI Nipus niger Casey STICHOLOTIDINAE SERANGIINI Delphastus catalinae (Horn) STICHOLOTIDINAE SERANGIINI Delphastus pusillus (LeConte) STICHOLOTIDINAE CEPHALOSCYMNINI Cephaloscymnus occidentalis Horn SCYMNINAE ZILINI Zilus aterrimus (Horn) SCYMNINAE ZILINI Zagloba ornata (Horn) SCYMNINAE STETHORINI Stethorus punctum picipes Casey SCYMNINAE SCYMNINI Cryptolaemus montrouzieri Mulsant SCYMNINAE SCYMNINI Didion punctatum (Melsheimer) SCYMNINAE SCYMNINI Didion longulum Casey SCYMNINAE SCYMNINI Scymnus nebulosus (LeConte) SCYMNINAE SCYMNINI Scymnus dificilis Casey SCYMNINAE SCYMNINI Scymnus fenderi Malkin SCYMNINAE SCYMNINI Scymnus caurinus Horn SCYMNINAE SCYMNINI Scymnus coniferarum Crotch SCYMNINAE SCYMNINI Scymnus pacificus Crotch SCYMNINAE SCYMNINI Scymnus pallens LeConte SCYMNINAE SCYMNINI Scymnus gilae Casey SCYMNINAE SCYMNINI Scymnus mimoides Gordon SCYMNINAE SCYMNINI Scymnus cervicalis Mulsant SCYMNINAE SCYMNINI Scymnus apithanus Gordon -

Emmetropization in Arthropods: a New Vision Test in Several
Emmetropization in arthropods: A new vision test in several arthropods suggests visual input may not be necessary to establish correct focusing A thesis submitted to the Graduate School of The University of Cincinnati (Department of Biological Sciences) In partial fulfillment of the requirements for the degree of Master of Science In the Department of Biological Sciences of the College of Arts and Sciences July 2019 By Madeline Owens B.S. Biological Sciences, University of Cincinnati (2016) Committee Chair: Dr. Elke K. Buschbeck, Ph.D. This work will be submitted to The Journal of Experimental Biology Authors: Madeline Owens, Elke K. Buschbeck ABSTRACT The visual systems of vertebrates and invertebrates alike must be able to achieve and maintain emmetropia, a state of correct focusing, in order to effectively execute visually guided behavior. Vertebrate studies, as well as a study in squid, have revealed that for them correct focusing is accomplished through a combination of gene regulation during early development, and homeostatic visual input from the environment that fine-tunes the eye. While eye growth towards emmetropia has been long researched in vertebrates, it is largely unknown how it is established in arthropods. To address this question, we built a micro-ophthalmoscope that allows us to directly measure how a lens projects an image onto the retina in the eyes of small, live arthropods, and to compare the eyes of different light-reared and dark-reared arthropods. Surprisingly, and in sharp contrast to vertebrates, our data on a diverse set of arthropods suggests that visual input in arthropods may not be necessary at least for the initial development of emmetropic eyes. -

Sibbald Point Provincial Park
SIBBALD POINT PROVINCIAL PARK One Malaise trap was deployed at Sibbald Point Provincial Park in 2014 (44.32982, -79.32737, 233m ASL; Figure 1). This trap collected arthropods for twenty weeks from May 1 – September 22, 2014. All 10 Malaise trap samples were processed using the bulk sample analysis protocol. A total of 1691 BINs were obtained. Over half the BINs captured were flies (Diptera), followed by bees, ants and wasps (Hymenoptera), beetles (Coleoptera), and moths and butterflies (Lepidoptera; Figure 2). In total, 399 arthropod species were named, representing 26.9% of the BINs from the site (Appendix 1). All the BINs were assigned at least to family, and 60.6% were assigned to a genus Figure 1. Malaise trap deployed at Sibbald Point (Appendix 2). Specimens collected from Sibbald Point Provincial Park in 2014. represent 160 different families and 473 genera. Figure 2. Taxonomy breakdown of BINs captured in the Malaise trap at Sibbald Point. APPENDIX 1. TAXONOMY REPORT Class Order Family Genus Species Arachnida Araneae Theridiidae Theridion Theridion murarium Diplopoda Julida Julidae Insecta Coleoptera Anthribidae Euxenus Euxenus punctatus Brentidae Betulapion Betulapion simile Byturidae Byturus Byturus unicolor Cantharidae Atalantycha Atalantycha bilineata Malthodes Malthodes pumilus Rhagonycha Rhagonycha fulva Carabidae Cicindela Cerambycidae Pidonia Pidonia ruficollis Urgleptes Urgleptes querci Chrysomelidae Altica Altica corni Phratora Phratora purpurea Phyllotreta Phyllotreta striolata Xanthonia Xanthonia decemnotata Cleridae -

Coleoptera: Coccinellidae): a Predator of Adelges Tsugae (Homoptera: Adelgidae)
Biology of Scymnus ningshanensis (Coleoptera: Coccinellidae): A Predator of Adelges tsugae (Homoptera: Adelgidae) Michael Montgomery, 1 Hongbin Wang,2 Defu Yao,2 Wenhau Lu,1 Nathan Havill,1 and Guangwu Li2 1 USDA Forest Service, Northeastern Center for Forest Health Research, 51 Millpond Road, Hamden, CT 06514 2 Chinese Academy of Forestry, Research Institute of Forest Environment and Protection Beijing 100091, People’s Republic of China Abstract Information is presented on the occurrence, development, and feeding of Scymnus (Neopullus) ningshanensis Yu et Yao. Information on its biology was collected in the field and laboratory in China and in quarantine in the United States. This lady beetle was found in China only on hemlock infested with Adelges tsugae Annand and did not occur on nearby pine infested with another adelgid. In the laboratory, newly hatched larvae survived only if they could feed on hemlock woolly adelgid eggs. Development of the immature stages was about 10% faster than S. (N.) sinuanodulus that occurs farther south in China. The climate in the native habitat of S. ningshanensis is similar to the climate of Connecticut in respect to temperature, but rainfall is lower in the winter and higher in the summer than in Connecticut. Eggs are laid in the spring for several weeks, both in the laboratory and field. Feeding on hemlock woolly adelgid eggs stimulates egg production. The species is univoltine and without a true diapause, although adults are inactive at temperatures below 7oC. Keywords: Lady beetles, biological control, bionomics, development. Introduction Scymnus (Neopullus) ningshanensis Yu et Yao is one of three Chinese lady beetles in the subgenus Neopullus imported into the USDA Forest Service Quarantine Laboratory, Ansonia, Connecticut, as a biological control agent for the hemlock woolly adelgid, Adelges tsugae Annand. -

Hemlock Woolly Adelgid Proceedings
Recognition of Imported Lady Beetles in the Tribe Scymnini Released in Eastern North America Lynn A. Jones1, Michael Montgomery1, Guoyue Yu 2, and Wenhau Lu3 1USDA Forest Service, Northeastern Center For Forest Health Research 51 Millpond Road, Hamden, CT 06514 2Institute of Plant and Environmental Protection, Beijing Academy of Agricultural and Forestry Science, Beijing, 100089 CHINA 3University of Rhode Island, Department of Plant Sciences Kingston, RI 02881 Abstract Adults of lady beetles in the tribe Scymnini imported for biological control of hemlock woolly adelgid, Adelges tsugae Annand, in eastern North America can be readily distinguished from native lady beetles (Coccinellidae). The imported lady beetles are in the genera Pseudoscymnus and Scymnus (Neopullus), both of which are Palearctic. These taxons have characteristics that can be used to separate them from Nearctic lady beetles. The most useful morphological feature is that the number of antennal segments is less than 11; ten-segmented in Scymnus (Neopullus) and nine- segmented in Pseudoscymnus. Elytral patterns and coloration also can be useful in field recognition. Larvae of the imported lady beetles are all in the tribe Scymnini, which can be distinguished from lady beetles in other tribes. A characteristic of the Scymnini is the wax covering of the last two larval instars and, for most species, the body lacks pronounced spines and is neither black or heavily sclerotized. Identification of a Scymnini larva to the genus level requires careful microscopic examination. Pseudoscymnus tsugae larval antennae have been described as one- segmented, but our examination indicates that they are two-segmented. Scymnus (Neopullus) larvae have three-segmented antennae. -

How Do Stemmata Grow? the Pursuit of Emmetropia in the Face Of
How do Stemmata Grow? The Pursuit of Emmetropia in the Face of Stepwise Growth A thesis submitted to the Graduate School of the University of Cincinnati (Division of Research and Advanced Studies of the University of Cincinnati) In partial fulfillment of the requirements for the degree of Master of Science In the Department of Biological Sciences of the College of Arts and Sciences 2014 By Shannon Werner B.A. Theater, Northern Kentucky University (2005) B.S. Neuroscience, University of Cincinnati (2011) Committee Chair: Dr Elke K. Buschbeck, Ph.D. i Abstract However complex or atypical the visual system, all of its components the lens, eye size and shape, retina and nervous system - must be well tuned to one another. As organisms grow, their eyes must be able to achieve and maintain emmetropia, a state where there is a good fit between the optical and receptor portions of the eye. Based on vertebrate studies, emmetropia is accomplished through a combination of initial regulation by genes, followed by homeostatic visual input from the environment for fine-tuning. How quickly and effectively emmetropia can be achieved will influence the ability of an animal to execute visually guided behavior. While there has been ample research into how vertebrates manage eye growth toward emmetropia, this has never been addressed in arthropods, which develop in a stepwise fashion through ecdysis. Of particular interest are larval holometabolous arthropods with camera type eyes, called stemmata in larval arthropods. Stemmata, such as those of the larval eyes of the Sunburst Diving Beetle (Thermonectus marmoratus, Coleoptera, Dytiscidae) must recover emmetropia after ecdysis, developing a well formed lens and receptor components that are well tuned to the lens’ optical properties. -

How to Look for Lady Beetles / Comment Trouver La Coccinelle
Field Guide LADY BEETLES of the Northwest Territories LES COCCINELLES des Territoires du Nord-Ouest Guide d’identification If you would like this information in another official language, call us. English Si vous voulez ces informations dans une autre langue officielle, contactez-nous. French Kīspin ki nitawihtīn ē nīhīyawihk ōma ācimōwin, tipwāsinān. Cree Tłı̨chǫ yatı k’ę̀ ę̀. Dı wegodı newǫ dè, gots’o gonede. Tłı̨chǫ Ɂerıhtł’ıś Dëne Sųłıné yatı t’a huts’elkër xa beyáyatı theɂą ɂat’e, nuwe ts’ën yółtı. Chipewyan Edı gondı dehgáh got’ı̨e zhatıé k’ę́ę́ edatł’éh enahddhę nıde naxets’ę́ edahłı.́ South Slavey K’áhshó got’ı̨ne xǝdǝ k’é hederı ɂedı̨htl’é yerınıwę nı ́dé dúle. North Slavey Jii gwandak izhii ginjìk vat’atr’ijąhch’uu zhit yinohthan jì’, diits’àt ginohkhìi. Gwich’in Uvanittuaq ilitchurisukupku Inuvialuktun, ququaqluta. Inuvialuktun ᑖᒃᑯᐊ ᑎᑎᕐᒃᑲᐃᑦ ᐱᔪᒪᒍᕕᒋᑦ ᐃᓄᒃᑎᑐᓕᕐᒃᓯᒪᓗᑎᒃ, ᐅᕙᑦᑎᓐᓄᑦ ᐅᖄᓚᔪᓐᓇᖅᑐᑎᑦ. Inuktitut Hapkua titiqqat pijumagupkit Inuinnaqtun, uvaptinnut hivajarlutit. Inuinnaqtun Aboriginal Languages Secretariat: 867-767-9346 ext. 71037 Francophone Affairs Secretariat: 867-767-9343 If you would like this information in another official language, call us. English Si vous voulez ces informations dans une autre langue officielle, contactez-nous. French Kīspin ki nitawihtīn ē nīhīyawihk ōma ācimōwin, tipwāsinān. Cree This identification guide includes all species of lady beetles (Family Coccinellidae) known to be Tłı̨chǫ yatı k’ę̀ ę̀. Dı wegodı newǫ dè, gots’o gonede. present in the Northwest Territories (NWT). Tłı̨chǫ ©Recommended 2018 Government citation: of the Northwest Territories Ɂerıhtł’ıś Dëne Sųłıné yatı t’a huts’elkër xa beyáyatı theɂą ɂat’e, nuwe ts’ën yółtı. -

Coleoptera: Coccinellidae) of Iowa, U.S.A
INSECTA MUNDI A Journal of World Insect Systematics 0091 An annotated checklist of the lady beetles (Coleoptera: Coccinellidae) of Iowa, U.S.A. Louis S. Hesler USDA Agricultural Research Service North Central Agricultural Research Laboratory 2923 Medary Avenue Brookings, SD 57006-9401, USA Date of Issue: September 25, 2009 CENTER FOR SYSTEMATIC ENTOMOLOGY, INC., Gainesville, FL Louis S. Hesler An annotated checklist of thelady beetles (Coleoptera: Coccinellidae) of Iowa, U.S.A. Insecta Mundi 0091: 1-10 Published in 2009 by Center for Systematic Entomology, Inc. P. O. Box 141874 Gainesville, FL 32614-1874 U. S. A. http://www.centerforsystematicentomology.org/ Insecta Mundi is a journal primarily devoted to insect systematics, but articles can be published on any non-marine arthropod taxon. Manuscripts considered for publication include, but are not limited to, systematic or taxonomic studies, revisions, nomenclatural changes, faunal studies, book reviews, phylo- genetic analyses, biological or behavioral studies, etc. Insecta Mundi is widely distributed, and refer- enced or abstracted by several sources including the Zoological Record, CAB Abstracts, etc. As of 2007, Insecta Mundi is published irregularly throughout the year, not as quarterly issues. As manuscripts are completed they are published and given an individual number. Manuscripts must be peer reviewed prior to submission, after which they are again reviewed by the editorial board to insure quality. One author of each submitted manuscript must be a current member of the Center for System- atic Entomology. Managing editor: Paul E. Skelley, e-mail: [email protected] Production editor: Michael C. Thomas, e-mail: [email protected] Editorial board: J. -

Insects Associated with Southern Magnolia (Magnolia Grandiflora L.) in East Tennessee
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 12-2002 Insects Associated with Southern Magnolia (Magnolia grandiflora L.) in East Tennessee Christopher Thomas Werle University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Entomology Commons Recommended Citation Werle, Christopher Thomas, "Insects Associated with Southern Magnolia (Magnolia grandiflora L.) in East Tennessee. " Master's Thesis, University of Tennessee, 2002. https://trace.tennessee.edu/utk_gradthes/2096 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Christopher Thomas Werle entitled "Insects Associated with Southern Magnolia (Magnolia grandiflora L.) in East Tennessee." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Entomology and Plant Pathology. Paris L. Lambdin, Major Professor We have read this thesis and recommend its acceptance: John A. Skinner, Jerome F. Grant Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) To the Graduate Council: I am submitting herewith a thesis written by Christopher Thomas Werle entitled “Insects Associated with Southern Magnolia (Magnolia grandiflora L.) in East Tennessee.” I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Master of Science, with a major in Entomology and Plant Pathology.