Orathecin (Rubitecan) Applicant: Eurogen Pharmaceuticals Ltd
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Options for the Treatment of Gemcitabine-Resistant Advanced Pancreatic Cancer
JOP. J Pancreas (Online) 2010 Mar 5; 11(2):113-123. REVIEW Options for the Treatment of Gemcitabine-Resistant Advanced Pancreatic Cancer Ioannis Gounaris, Kamarul Zaki, Pippa Corrie Oncology Centre, Cambridge University Hospitals NHS Trust. Cambridge, United Kingdom Summary Context Pancreatic cancer is noteworthy in that the number of patients dying from the disease is roughly equal to the number diagnosed. For more than a decade, gemcitabine has constituted the standard of care for the palliative treatment of the majority of patients who present with metastatic or relapsed disease, although the survival gains are limited. Despite a median survival of less than 6 months, there is a significant proportion of advanced pancreatic cancer patients who progress on gemcitabine that remains fit and these patients are candidates for second-line treatment. Methods The OVID MEDLINE database was searched from 1950 to present using the MeSH terms “pancreatic neoplasms”, “drug treatment” and “gemcitabine”. After excluding non-relevant results, 31 published studies were identified. These results were supplemented by searching the last three (2007-2009) American Society of Clinical Oncology (ASCO) Proceedings of Annual Meetings for studies published only in abstract form and reviewing reference lists of published articles. Results and discussion The evidence for second line treatments of metastatic pancreatic cancer consists mostly of single arm, small phase II studies. Oxaliplatin-fluoropyrimidine combinations appear promising and have shown increased survival compared to best supportive care. As the molecular pathways governing pancreatic cancer are unravelled, novel targeted therapies may offer the greatest promise for this disease either given alone, combined with one another, or with cytotoxic agents. -

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

Identification of Repurposed Drugs for Chordoma Therapy
Identification of Repurposed Drugs for Chordoma Therapy. Menghang Xia, Ph.D. Division of Pre-Clinical Innovation National Center for Advancing Translational Sciences National Institutes of Health Fourth International Chordoma Research Workshop Boston, March 22, 2013 NIH Chemical Genomics Center Founded in 2004 • National Center for Advancing Translational Sciences (NCATS) • >100 staff: Biologists, Chemists, Informatics and Engineers Robotic HTS facility Mission • Development of chemical probes for novel biology • Novel targets, rare/neglected diseases • New technologies/paradigms for assay development, screening, informatics, chemistry Collaborations • >200 investigators worldwide • 60% NIH extramural • 25% NIH intramural • 15% Foundations, Research Consortia, Pharma/Biotech Steps in the drug development process Make Create Test modifications Test in Test in testing >100,000 to active animals for humans for system chemicals for chemicals to safety, safety, (aka, activity on make suitable effectiveness effectiveness “assay”) target for human use Two approaches to therapeutics for rare and neglected diseases 1-2 years? >400,000 compounds, 15 yrs Lead Preclinical Clinical Screen Hit Lead Optimization Development Trials 3500 drugs The NCGC Pharmaceutical Collection Procurement in Drug Source In house process Total US FDA* 1635 182 1817 UK/EU/Canada/Japan 756 177 933 Total Approved 2391 359 2750 Investigational 928 3953 4881 Total 3319 4312 7631 * These counts include approved veterinary drugs Informatics sources for NPC o US FDA: Orange Book, OTC, NDC, Green Book, Drugs at FDA o Britain NHS o EMEA o Health Canada o Japan NHI Physical sources for NPC o Procurement from >70 suppliers worldwide o In-house purification of APIs from marketed forms Drug plate composition o Synthesis Comprehensive Drug Repurposing Library Access to the NPC (http://tripod.nih.gov/npc/) Chordoma Screening Project • Cell lines Chordoma cell lines screened: U-CH1 and U-CH2B . -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Research Paper Human Multidrug Resistance Associated Protein 4
Pharmaceutical Research ( # 2005) DOI: 10.1007/s11095-005-7595-9 http://www.paper.edu.cn Research Paper Human Multidrug Resistance Associated Protein 4 Confers Resistance to Camptothecins Quan Tian,1 Jing Zhang,1 Theresa May Chin Tan,2 Eli Chan,1 Wei Duan,2 Sui Yung Chan,1 Urs Alex Boelsterli,1,3 Paul Chi-Lui Ho,1 Hongyuan Yang,2 Jin-Song Bian,3 Min Huang,4 Yi-Zhun Zhu,3 Weiping Xiong,5 Xiaotian Li,6 and Shufeng Zhou1,7 Received May 6, 2005; accepted July 22, 2005 Purpose. The multidrug resistance associated protein (MRP) 4 is a member of the adenosine triphosphate (ATP)-binding cassette transporter family. Camptothecins (CPTs) have shown substantial anticancer activity against a broad spectrum of tumors by inhibiting DNA topoisomerase I, but tumor resistance is one of the major reasons for therapeutic failure. P-glycoprotein, breast cancer resistance protein, MRP1, and MRP2 have been implicated in resistance to various CPTs including CPT-11 (irinotecan), SN-38 (the active metabolite of CPT-11), and topotecan. In this study, we explored the resistance profiles and intracellular accumulation of a panel of CPTs including CPT, CPT-11, SN-38, rubitecan, and 10-hydroxy-CPT (10-OH-CPT) in HepG2 cells with stably overexpressed human MRP4. Other anticancer agents such as paclitaxel, cyclophosphamide, and carboplatin were also included. Methods. HepG2 cells were transfected with an empty vehicle plasmid (V/HepG2) or human MRP4 (MRP4/HepG2). The resistance profiles of test drugs in exponentially growing V/HepG2 and MRP4/ HepG2 cells were examined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazonium bromide (MTT) assay with 4 or 48 h exposure time of the test drug in the absence or presence of various MRP4 inhibitors. -
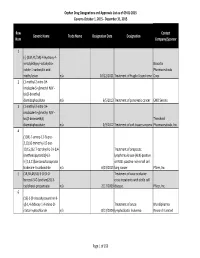
Orphan Drug Designation List
Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 1 (‐)‐(3aR,4S,7aR)‐4‐Hydroxy‐4‐ m‐tolylethynyl‐octahydro‐ Novartis indole‐1‐carboxylic acid Pharmaceuticals methyl ester n/a 10/12/2011 Treatment of Fragile X syndrome Corp. 2 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐broethyl) diamidophosphate n/a 6/5/2013 Treatment of pancreatic cancer EMD Serono 3 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐bromoethyl) Threshold diamidophosphate n/a 3/9/2012 Treatment of soft tissue sarcoma Pharmaceuticals, Inc. 4 (1OR)‐7‐amino‐12‐fluoro‐ 2,10,16‐trimethyl‐15 oxo‐ 10,15,16,17‐tetrahydro‐2H‐8,4‐ Treatment of anaplastic (metheno)pyrazolo[4,3‐ lymphoma kinase (ALK)‐positive h][2,5,11]benzoxadiazacyclote or ROS1‐positive non‐small cell tradecine‐3‐carbonitrile n/a 6/23/2015 lung cancer Pfizer, Inc. 5 (1R,3R,4R,5S)‐3‐O‐[2‐O‐ Treatment of vaso‐occlusive benzoyl‐3‐O‐(sodium(2S)‐3‐ crisis in patients with sickle cell cyclohexyl‐propanoate‐ n/a 2/17/2009 disease. Pfizer, Inc. 6 (1S)‐1‐(9‐deazahypoxanthin‐9‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ Treatment of acute Mundipharma ribitol‐hydrochloride n/a 8/13/2004 lymphoblastic leukemia Research Limited Page 1 of 359 Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 7 Treatment of chronic lymphocytic leukemia and related leukemias to include (1S)‐1‐(9‐deazahypoxanthin‐9‐ prolymphocytic leukemia, adult T‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ cell leukemia, and hairy cell Mundipharma ribitol‐hydrochloride n/a 8/10/2004 leukemia Research Ltd. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0226431 A1 Habib (43) Pub
US 20090226431A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0226431 A1 Habib (43) Pub. Date: Sep. 10, 2009 (54) TREATMENT OF CANCER AND OTHER Publication Classification DISEASES (51) Int. Cl. A 6LX 3/575 (2006.01) (76)76) InventorInventor: Nabilabil Habib,Habib. Beirut (LB(LB) C07J 9/00 (2006.01) Correspondence Address: A 6LX 39/395 (2006.01) 101 FEDERAL STREET A6IP 29/00 (2006.01) A6IP35/00 (2006.01) (21) Appl. No.: 12/085,892 A6IP37/00 (2006.01) 1-1. (52) U.S. Cl. ...................... 424/133.1:552/551; 514/182: (22) PCT Filed: Nov.30, 2006 514/171 (86). PCT No.: PCT/US2O06/045665 (57) ABSTRACT .."St. Mar. 6, 2009 The present invention relates to a novel compound (e.g., 24-ethyl-cholestane-3B.5C,6C.-triol), its production, its use, and to methods of treating neoplasms and other tumors as Related U.S. Application Data well as other diseases including hypercholesterolemia, (60) Provisional application No. 60/741,725, filed on Dec. autoimmune diseases, viral diseases (e.g., hepatitis B, hepa 2, 2005. titis C, or HIV), and diabetes. F2: . - 2 . : F2z "..., . Cz: ".. .. 2. , tie - . 2 2. , "Sphagoshgelin , , re Cls Phosphatidiglethanolamine * - 2 .- . t - r y ... CBs .. A . - . Patent Application Publication Sep. 10, 2009 Sheet 1 of 16 US 2009/0226431 A1 E. e'' . Phosphatidylcholine. " . Ez'.. C.2 . Phosphatidylserias. * . - A. z' C. w E. a...2 .". is 2 - - " - B 2. Sphingoshgelin . Cls Phosphatidglethanglamine Figure 1 Patent Application Publication Sep. 10, 2009 Sheet 2 of 16 US 2009/0226431 A1 Chile Phosphater Glycerol Phosphatidylcholine E. -

Chemotherapy and Radiotherapy for Advanced Pancreatic Cancer (Review)
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Enlighten Cochrane Database of Systematic Reviews Chemotherapy and radiotherapy for advanced pancreatic cancer (Review) Chin V, Nagrial A, Sjoquist K, O’Connor CA, Chantrill L, Biankin AV, Scholten RJPM, Yip D Chin V, Nagrial A, Sjoquist K, O’Connor CA, Chantrill L, Biankin AV, Scholten RJPM, Yip D. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Database of Systematic Reviews 2018, Issue 3. Art. No.: CD011044. DOI: 10.1002/14651858.CD011044.pub2. www.cochranelibrary.com Chemotherapy and radiotherapy for advanced pancreatic cancer (Review) Copyright © 2018 The Cochrane Collaboration. Published by John Wiley & Sons, Ltd. TABLE OF CONTENTS HEADER....................................... 1 ABSTRACT ...................................... 1 PLAINLANGUAGESUMMARY . 2 SUMMARY OF FINDINGS FOR THE MAIN COMPARISON . ..... 4 BACKGROUND .................................... 6 OBJECTIVES ..................................... 7 METHODS ...................................... 7 RESULTS....................................... 10 Figure1. ..................................... 11 Figure2. ..................................... 15 Figure3. ..................................... 16 ADDITIONALSUMMARYOFFINDINGS . 22 DISCUSSION ..................................... 28 AUTHORS’CONCLUSIONS . 30 ACKNOWLEDGEMENTS . 31 REFERENCES ..................................... 31 CHARACTERISTICSOFSTUDIES . 45 DATAANDANALYSES. 112 Analysis 1.1. Comparison 1 Anti-cancer -

Antitumors for Research and Experimental Use
Antitumors for Research and Experimental Use Platinum-containing Antitumor Agents Hormonal Antitumor Agents C2043 Carboplatin 100mg / 1g A1947 DL-Aminoglutethimide 5g / 25g D3371 Cisplatin 100mg / 1g New A2370 Anastrozole 100mg O0372 Oxaliplatin 100mg New B3206 Bicalutamide 200mg / 1g D1961 Dexamethasone 1g Antitumor Antimetabolites New E0941 Exemestane 200mg / 1g A0907 Allopurinol 25g / 250g F0663 Flutamide 5g / 25g New A2528 Acadesine 50mg New L0248 Letrozole 1g New A2033 Ladakamycin 100mg / 1g New L0249 Leuprorelin Acetate 25mg A2232 Decitabine 20mg / 100mg M1964 Medroxyprogesterone Acetate New C2663 Carmofur 5g / 25g 1g / 5g New C2499 Cladribine 50mg M1949 Megestrol Acetate 1g / 5g C2500 Clofarabine 20mg / 100mg P0637 Prednisolone 1g / 5g / 25g New D4342 5'-Deoxy-5-uorocytidine 1g / 5g P1276 Prednisone 5g / 25g F0151 5-Fluorouracil 5g / 25g New R0109 Raloxifene Hydrochloride 1g G0367 Gemcitabine Hydrochloride T2510 Tamoxifen Citrate 1g / 5g 100mg / 1g New T2832 Toremifene Citrate 1g / 5g H0310 Hydroxyurea 5g / 25g M0063 6-Mercaptopurine Monohydrate Antitumor Alkylating Agents 1g / 5g New B4033 Bendamustine Hydrochloride M1664 Methotrexate Hydrate 1g / 5g Hydrate 200mg F0635 Tegafur 5g / 25g B1022 Busulfan 25g T0212 6-Thioguanine 1g / 5g New C2634 Carmustine 100mg V0098 Vidarabine Monohydrate 1g / 5g C2236 Cyclophosphamide Monohydrate 5g / 25g Antitumor Plant Alkaloids and Terpenoids D3634 Dacarbazine 1g / 5g A2063 9-Aminocamptothecin 10mg New L0251 Lomustine 200mg / 1g C1495 (S)-(+)-Camptothecin 100mg / 1g N0821 Nimustine Hydrochloride -

Vivacitas Oncology Appoints Mr. Heng Fai Chan to the Board of Directors
Vivacitas Oncology Appoints Mr. Heng Fai Chan to the Board of Directors Walnut Creek, CA – May 23rd, 2017 – Vivacitas Oncology Inc. (“Vivacitas”, or the “Company”), a biopharmaceutical company focused on developing camptothecin-based medications for cancer patients, announces the appointment of Mr. Heng Fai Chan to the Company’s Board of Directors. Concurrently, Heng Fai Enterprises Pte Ltd., controlled by Mr. Chan, made an investment into Vivacitas Oncology through the purchase of common stock. Mr. Chan brings over 40 years of international business experience to the board of directors with an exemplary track record of restructuring companies. “Vivacitas represents a tremendous investment opportunity in the field of oncologic drug development as the pipeline assets have progressed to a relatively late stage and are differentiated from other camptothecins in their improved formulations and reduced side effects,” said Mr. Chan. “Led by the Company’s scientific founder, the late Dr. Joseph Rubinfeld, the acquisition of Rubitecan (Orathecin) in the fourth quarter of 2016 enhanced the Company’s pipeline by adding a unique camptothecin-based formula that can be given orally. Taken together with the pipeline drug AR-67, acquired from a public company in early 2016, Vivacitas’ mid-to late- stage portfolio is positioned to fight a spectrum of cancers using camptothecins, which have been known for decades to have chemotherapeutics effects in a multitude of cancers. My immediate focus is to help the Company with its capital raising efforts and strategic transactions as we bring new treatments forward for patients.” Mr. Chan, based in Singapore, brings more than 40 years of operational experience; from founding companies to restructuring others globally, his companies speak to his expertise. -

Discovery of Anti-Amoebic Inhibitors from Screening the MMV Pandemic Response Box on Balamuthia Mandrillaris, Naegleria Fowleri, and Acanthamoeba Castellanii
pathogens Article Discovery of Anti-Amoebic Inhibitors from Screening the MMV Pandemic Response Box on Balamuthia mandrillaris, Naegleria fowleri, and Acanthamoeba castellanii 1,2, , , 2,3, 2,3, Christopher A. Rice * y z , Emma V. Troth y , A. Cassiopeia Russell y and Dennis E. Kyle 1,2,3,* 1 Department of Cellular Biology, University of Georgia, Athens, GA 30602, USA 2 Center for Tropical and Emerging Global Diseases, Athens, GA 30602, USA; [email protected] (E.V.T.); [email protected] (A.C.R.) 3 Department of Infectious Diseases, University of Georgia, Athens, GA 30602, USA * Correspondence: [email protected] (C.A.R.); [email protected] (D.E.K.) These authors contributed equally to this work. y Current address: Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of z Georgia, Athens, GA 30602, USA. Received: 12 May 2020; Accepted: 9 June 2020; Published: 16 June 2020 Abstract: Pathogenic free-living amoebae, Balamuthia mandrillaris, Naegleria fowleri, and several Acanthamoeba species are the etiological agents of severe brain diseases, with case mortality rates > 90%. A number of constraints including misdiagnosis and partially effective treatments lead to these high fatality rates. The unmet medical need is for rapidly acting, highly potent new drugs to reduce these alarming mortality rates. Herein, we report the discovery of new drugs as potential anti-amoebic agents. We used the CellTiter-Glo 2.0 high-throughput screening methods to screen the Medicines for Malaria Ventures (MMV) Pandemic Response Box in a search for new active chemical scaffolds. Initially, we screened the library as a single-point assay at 10 and 1 µM. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.