Initial Genome Sequencing and Analysis of Multiple Myeloma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BCL7A As a Novel Prognostic Biomarker for Glioma Patients
BCL7A as a novel prognostic biomarker for glioma patients Junhui Liu ( [email protected] ) Renmin Hospital of Wuhan University: Wuhan University Renmin Hospital Lun Gao Renmin Hospital of Wuhan University: Wuhan University Renmin Hospital Baowei Ji Renmin Hospital of Wuhan University: Wuhan University Renmin Hospital Rongxin Geng Renmin Hospital of Wuhan University: Wuhan University Renmin Hospital Jing Chen Renmin Hospital of Wuhan University: Wuhan University Renmin Hospital Xiang Tao Renmin Hospital of Wuhan University: Wuhan University Renmin Hospital Qiang Cai Renmin Hospital of Wuhan University: Wuhan University Renmin Hospital Zhibiao Chen Renmin Hospital of Wuhan University: Wuhan University Renmin Hospital Research Article Keywords: BCL7 family, glioma, prognosis, immune, Temozolomide Posted Date: April 21st, 2021 DOI: https://doi.org/10.21203/rs.3.rs-390165/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Journal of Translational Medicine on August 6th, 2021. See the published version at https://doi.org/10.1186/s12967-021-03003-0. Page 1/24 Abstract Background: Glioma is the most common primary brain tumor and represents one of the most aggressive and lethal types of human cancer. BCL7 family has been found in several cancer types and could be involved in tumor progression. While the role of BCL7 family in human glioma has remained to be elucidated. Methods: Paran-embedded tumor samples were obtained to detect BCL7 expression by performing in glioma. Data (including normalized gene expression and corresponding clinical data) were obtained from Gliovis, CGGA, GEO, cBioportal and Oncomine and were used to investigate BCL7 genes expression in glioma. -

Integrating Genomic Alterations in Diffuse Large B-Cell Lymphoma Identifies New Relevant Pathways and Potential Therapeutic Targets
OPEN Leukemia (2018) 32, 675–684 www.nature.com/leu ORIGINAL ARTICLE Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets K Karube1,2, A Enjuanes1,3, I Dlouhy1, P Jares1,3, D Martin-Garcia1,3, F Nadeu1,3, GR Ordóñez4, J Rovira1, G Clot1,3, C Royo1, A Navarro1,3, B Gonzalez-Farre1,3, A Vaghefi1, G Castellano1, C Rubio-Perez5, D Tamborero5, J Briones6, A Salar7, JM Sancho8, S Mercadal9, E Gonzalez-Barca9, L Escoda10, H Miyoshi11, K Ohshima11, K Miyawaki12, K Kato12, K Akashi12, A Mozos13, L Colomo1,7, M Alcoceba3,14, A Valera1, A Carrió1,3, D Costa1,3, N Lopez-Bigas5,15, R Schmitz16, LM Staudt16, I Salaverria1,3, A López-Guillermo1,3 and E Campo1,3 Genome studies of diffuse large B-cell lymphoma (DLBCL) have revealed a large number of somatic mutations and structural alterations. However, the clinical significance of these alterations is still not well defined. In this study, we have integrated the analysis of targeted next-generation sequencing of 106 genes and genomic copy number alterations (CNA) in 150 DLBCL. The clinically significant findings were validated in an independent cohort of 111 patients. Germinal center B-cell and activated B-cell DLBCL had a differential profile of mutations, altered pathogenic pathways and CNA. Mutations in genes of the NOTCH pathway and tumor suppressor genes (TP53/CDKN2A), but not individual genes, conferred an unfavorable prognosis, confirmed in the independent validation cohort. A gene expression profiling analysis showed that tumors with NOTCH pathway mutations had a significant modulation of downstream target genes, emphasizing the relevance of this pathway in DLBCL. -

Pathognomonic and Epistatic Genetic Alterations in B-Cell Non-Hodgkin
bioRxiv preprint doi: https://doi.org/10.1101/674259; this version posted June 19, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Pathognomonic and epistatic genetic alterations in 2 B-cell non-Hodgkin lymphoma 3 4 Man Chun John Ma1¥, Saber Tadros1¥, Alyssa Bouska2, Tayla B. Heavican2, Haopeng Yang1, 5 Qing Deng1, Dalia Moore3, Ariz Akhter4, Keenan Hartert3, Neeraj Jain1, Jordan Showell1, 6 Sreejoyee Ghosh1, Lesley Street5, Marta Davidson5, Christopher Carey6, Joshua Tobin7, 7 Deepak Perumal8, Julie M. Vose9, Matthew A. Lunning9, Aliyah R. Sohani10, Benjamin J. 8 Chen11, Shannon Buckley12, Loretta J. Nastoupil1, R. Eric Davis1, Jason R. Westin1, Nathan H. 9 Fowler1, Samir Parekh8, Maher K. Gandhi7, Sattva S. Neelapu1, Douglas Stewart5, Javeed 10 Iqbal2, Timothy Greiner2, Scott J. Rodig13, Adnan Mansoor5, Michael R. Green1,14,15* 11 1Department of Lymphoma and Myeloma, Division of Cancer Medicine, The University of Texas MD 12 Anderson Cancer Center, Houston, TX, USA; 2Department of Pathology and Microbiology, University of 13 Nebraska Medical Center, Omaha, NE, USA; 3Eppley Institute for Research in Cancer and Allied 14 Diseases, University of Nebraska Medical Center, Omaha, NE, USA; 4Department of Pathology and 15 Laboratory Medicine, University of Calgary, Calgary, AB, Canada; 5Section of Hematology, Department of 16 Medicine, University -

BCL7A Antibody Cat
BCL7A Antibody Cat. No.: 45-325 BCL7A Antibody 45-325 Immunofluorescence analysis of 45-325 Flow cytometric analysis of paraformaldehyde paraformaldehyde fixed U2OS cells, permeabilized with fixed Daudi cells (blue line), permeabilized with 0.5% 0.15% Triton. Primary incubation 1hr (10ug/ml) followed Triton. Primary incubation 1hr (10ug/ml) followed by Alexa by Alexa Fluor 488 secondary antibody (2ug/ml), showing Fluor 488 secondary antibody (1ug/ml). IgG control: nuclear staining. Actin filaments were stained wit Unimmunized goat IgG (black line) fo Specifications HOST SPECIES: Goat SPECIES REACTIVITY: Human HOMOLOGY: Expected Species Reactivity based on sequence homology: Mouse, Rat, Dog, Cow, Pig IMMUNOGEN: The immunogen for this antibody is: C-MKLEASQQNSEEM TESTED APPLICATIONS: ELISA, Flow, IF, WB October 2, 2021 1 https://www.prosci-inc.com/bcl7a-antibody-45-325.html Peptide ELISA: antibody detection limit dilution 1:4000.Western Blot:Approx 26kDa band observed in Human Lymph Node lysates (calculated MW of 25.0kDa according to NP_066273.1). Recommended concentration: 1-3ug/ml. Primary incubation 1 hour at APPLICATIONS: room temperature.Immunofluorescence: Strong expression of the protein seen in the nuclei of U2OS cells. Recommended concentration: 10ug/ml. Flow Cytometry: Flow cytometric analysis of Daudi cells. Recommended concentration: 10ug/ml. This antibody is expected to recognize both reported isoforms (NP_066273.1; SPECIFICITY: NP_001019979.1). POSITIVE CONTROL: 1) Cat. No. 1201 - HeLa Cell Lysate Properties Purified from goat serum by ammonium sulphate precipitation followed by antigen PURIFICATION: affinity chromatography using the immunizing peptide. CLONALITY: Polyclonal CONJUGATE: Unconjugated PHYSICAL STATE: Liquid Supplied at 0.5 mg/ml in Tris saline, 0.02% sodium azide, pH7.3 with 0.5% bovine serum BUFFER: albumin. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

Nonimmunoglobulin Target Loci of Activation-Induced Cytidine Deaminase (AID) Share Unique Features with Immunoglobulin Genes
Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes Lucia Katoa, Nasim A. Beguma, A. Maxwell Burroughsb, Tomomitsu Doia,1, Jun Kawaib, Carsten O. Daubb, Takahisa Kawaguchic, Fumihiko Matsudac, Yoshihide Hayashizakib, and Tasuku Honjoa,2 aDepartment of Immunology and Genomic Medicine and cThe Center for Genomic Medicine, Graduate School of Medicine, Kyoto University, Kyoto 606-8501, Japan; and bRIKEN Omics Science Center (OSC), RIKEN Yokohama Institute, Yokohama, Kanagawa 230-0045, Japan Contributed by Tasuku Honjo, December 28, 2011 (sent for review December 5, 2011) Activation-induced cytidine deaminase (AID) is required for both (19, 20). Non-B structure involvement has recently been repor- somatic hypermutation and class-switch recombination in activated ted in transcription-associated mutations in repetitive sequences B cells. AID is also known to target nonimmunoglobulin genes and such as the dinucleotide repeat hot spots or triplet repeat ex- introduce mutations or chromosomal translocations, eventually pansion/contractions causing Huntington’s disease (17, 21, 22). causing tumors. To identify as-yet-unknown AID targets, we AID-dependent DNA cleavage is, in general, specific to the Ig screened early AID-induced DNA breaks by using two independent locus. However, a number of reports have shown that AID can genome-wide approaches. Along with known AID targets, this induce DNA cleavage in non-Ig loci. AID non-Ig targets were screen identified a set of unique genes (SNHG3, MALAT1, BCL7A, first demonstrated by studies on AID transgenic mice that pro- and CUX1) and confirmed that these loci accumulated mutations as duce numerous T lymphomas, in which vast numbers of muta- frequently as Ig locus after AID activation. -

Next Generation Sequencing: Advances in Characterizing the Methylome
Genes 2010, 1, 143-165; doi:10.3390/genes1020143 OPEN ACCESS genes ISSN 2073-4425 www.mdpi.com/journal/genes Review Next Generation Sequencing: Advances in Characterizing the Methylome Kristen H. Taylor 1, Huidong Shi 2 and Charles W. Caldwell 1,* 1 University of Missouri-Columbia School of Medicine, Ellis Fischel Cancer Center, Columbia, MO 65212, USA; E-Mail: [email protected] 2 Medical College of Georgia, Augusta, GA 30912, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-573-882-1283; Fax: +1-573-884-4612. Received: 3 May 2010; in revised form: 22 June 2010 / Accepted: 28 June 2010 / Published: 1 July 2010 Abstract: Epigenetic modifications play an important role in lymphoid malignancies. This has been evidenced by the large body of work published using microarray technologies to generate methylation profiles for numerous types and subtypes of lymphoma and leukemia. These studies have shown the importance of defining the epigenome so that we can better understand the biology of lymphoma. Recent advances in DNA sequencing technology have transformed the landscape of epigenomic analysis as we now have the ability to characterize the genome-wide distribution of chromatin modifications and DNA methylation using next-generation sequencing. To take full advantage of the throughput of next-generation sequencing, there are many methodologies that have been developed and many more that are currently being developed. Choosing the appropriate methodology is fundamental to the outcome of next-generation sequencing studies. In this review, published technologies and methodologies applicable to studying the methylome are presented. -

Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development
International Journal of Molecular Sciences Article Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development Nashwa El Hadidy 1 and Vladimir N. Uversky 1,2,* 1 Department of Molecular Medicine, Morsani College of Medicine, University of South Florida, 12901 Bruce B. Downs Blvd. MDC07, Tampa, FL 33612, USA; [email protected] 2 Laboratory of New Methods in Biology, Institute for Biological Instrumentation of the Russian Academy of Sciences, Federal Research Center “Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences”, Pushchino, 142290 Moscow Region, Russia * Correspondence: [email protected]; Tel.: +1-813-974-5816; Fax: +1-813-974-7357 Received: 20 September 2019; Accepted: 21 October 2019; Published: 23 October 2019 Abstract: The two-meter-long DNA is compressed into chromatin in the nucleus of every cell, which serves as a significant barrier to transcription. Therefore, for processes such as replication and transcription to occur, the highly compacted chromatin must be relaxed, and the processes required for chromatin reorganization for the aim of replication or transcription are controlled by ATP-dependent nucleosome remodelers. One of the most highly studied remodelers of this kind is the BRG1- or BRM-associated factor complex (BAF complex, also known as SWItch/sucrose non-fermentable (SWI/SNF) complex), which is crucial for the regulation of gene expression and differentiation in eukaryotes. Chromatin remodeling complex BAF is characterized by a highly polymorphic structure, containing from four to 17 subunits encoded by 29 genes. The aim of this paper is to provide an overview of the role of BAF complex in chromatin remodeling and also to use literature mining and a set of computational and bioinformatics tools to analyze structural properties, intrinsic disorder predisposition, and functionalities of its subunits, along with the description of the relations of different BAF complex subunits to the pathogenesis of various human diseases. -

Mutational Landscape of High-Grade B-Cell Lymphoma with MYC-, BCL2 And/Or
medRxiv preprint doi: https://doi.org/10.1101/2021.07.13.21260465; this version posted July 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. 1 Title: Mutational landscape of high-grade B-cell lymphoma with MYC-, BCL2 and/or 2 BCL6 rearrangements characterized by whole-exome sequencing 3 4 Authors: Axel Künstner1,2,3*, Hanno M. Witte4,5*, Jörg Riedl4,6*, Veronica Bernard6, 5 Stephanie Stölting6, Hartmut Merz6, Vito Olschewski4, Wolfgang Peter7,8, Julius Ketzer9, 6 Yannik Busch7, Peter Trojok7, Nikolas von Bubnoff3,4, Hauke Busch1,2,3**, Alfred C. Feller6**, 7 Niklas Gebauer3,4** 8 9 Affiliations: 10 1 Medical Systems Biology Group, University of Lübeck, Ratzeburger Allee 160, 23538 11 Lübeck, Germany 12 2 Institute for Cardiogenetics, University of Lübeck, Ratzeburger Allee 160, 23538 Lübeck, 13 Germany 14 3 University Cancer Center Schleswig-Holstein, University Hospital of Schleswig- Holstein, 15 Campus Lübeck, 23538 Lübeck, Germany 16 4 Department of Hematology and Oncology, University Hospital of Schleswig-Holstein, 17 Campus Lübeck, Ratzeburger Allee 160, 23538 Lübeck, Germany 18 5 Department of Hematology and Oncology, Federal Armed Forces Hospital Ulm, Oberer 19 Eselsberg 40, 89081 Ulm 20 6 Hämatopathologie Lübeck, Reference Centre for Lymph Node Pathology and 21 Hematopathology, Lübeck, Germany 22 7 HLA Typing Laboratory of the Stefan-Morsch-Foundation, 557565 Birkenfeld, Germany 23 8 Institut für Tranfusionsmedizin, Universitätsklinikum Köln. Kerpenerstr. 62, 50937 Köln 24 9 Department of Paediatrics, University Hospital of Schleswig-Holstein, Campus Luebeck, 25 23538 Luebeck, Germany 26 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice.1 medRxiv preprint doi: https://doi.org/10.1101/2021.07.13.21260465; this version posted July 16, 2021. -

Original Article BCL7B, a Predictor of Poor Prognosis of Pancreatic Cancers, Promotes Cell Motility and Invasion by Influencing CREB Signaling
Am J Cancer Res 2018;8(3):387-404 www.ajcr.us /ISSN:2156-6976/ajcr0070155 Original Article BCL7B, a predictor of poor prognosis of pancreatic cancers, promotes cell motility and invasion by influencing CREB signaling Keisuke Taniuchi1,2, Mutsuo Furihata3, Seiji Naganuma3, Ken Dabanaka4, Kazuhiro Hanazaki4, Toshiji Saibara1,2 Departments of 1Gastroenterology and Hepatology, 2Endoscopic Diagnostics and Therapeutics, 3Pathology, 4Sur- gery, Kochi Medical School, Kochi University, Nankoku, Kochi 783-8505, Japan Received December 1, 2017; Accepted December 12, 2017; Epub March 1, 2018; Published March 15, 2018 Abstract: The functions of B-cell CLL/lymphoma 7B (BCL7B) are unknown and the protein lacks any known func- tional domains. The aim of this study was to investigate the role of BCL7B in the motility and invasiveness of pan- creatic cancer cells. Immunohistochemistry was performed to determine whether high BCL7B expression in human pancreatic cancer tissues is correlated with poor prognosis. High BCL7B expression was an independent predictor of worse overall survival of pancreatic cancer patients. Immunocytochemistry showed that BCL7B was accumulated in cell protrusions of migrating pancreatic cancer cells. Knockdown of BCL7B inhibited the motility and invasiveness of pancreatic cancer cells through a decrease in cell protrusions. Phosphoprotein array analysis was performed to determine BCL7B-associated intracellular signaling pathways. Suppression of BCL7B increased phosphorylated CREB expression in pancreatic cancer cells, and knockdown of CREB promoted the motility and invasiveness by increasing cell protrusions. The combined data suggest that BCL7B promotes pancreatic cancer cell motility and invasion through a signaling pathway that involves dephosphorylation of CREB. Keywords: BCL7B, pancreatic cancer, cell invasion, CREB, actin-cytoskeleton Introduction tion syndrome [3]. -
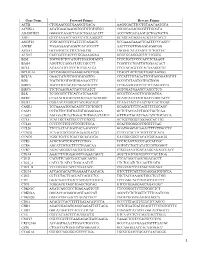
Gene Name Forward Primer Reverse Primer ACTB
Gene Name Forward Primer Reverse Primer ACTB CTGGAACGGTGAAGGTGACA AAGGGACTTCCTGTAACAATGCA ACVRL1 ACATGAAGAAGGTGGTGTGTGTGG CGGGCAGAGGGGTTTGGGTA ADAMDEC1 GGGGCCAGACTACACTGAAACATT ACCCGTCACAAGTACTGATGCTG AHI1 GTCCAAAACTACCCCATCAAGGCT GCAGCACAGGAACGTATCACCT ANGPT2 TGGCAGCGTTGATTTTCAGAGG GCGAAACAAACTCATTTCCCAGCC ANPEP TGAAGAAGCAGGTCACACCCCT AACTCCGTTGGAGCAGGCGG APOA1 GCCGTGCTCTTCCTGACGG TGGGACACATAGTCTCTGCCGC ATXN7 CACCGCCCACTCTGGAAAAGAA GGGTGCAGGGCTTCTTGGTG B2M TGCTGTCTCCATGTTTGATGTATCT TCTCTGCTCCCCACCTCTAAGT BAG4 AGGTTCCAGGATATCCGCCTT TCGGTCCTGATTGTGGAACACT BCL2 ACAACATCGCCCTGTGGATGA CCGTACAGTTCCACAAAGGCAT BCL2L14 GCTCAGGGTCAAAGGACGTTGG TCAGCTACTCGGTTGGCAATGG BCL7A GAACCATGTCGGGCAGGTCG CCCATTTGTAGATTCGTAGGGATGTGT BIN1 TGCTGTCGTGGTGGAGACCTTC GCCGTGTAGTCGTGCTGGG BIRC3 TGCTATCCACATCAGACAGCCC TCTGAATGGTCTTCTCCAGGTTCA BIRC5 TTCTCAAGGACCACCGCATCT AGTGGATGAAGCCAGCCTCG BLK TCGGGGTCTTCACCATCAAAGC GCGCTCCAGGTTGCGGATGA BTRC CCAAATGTGTCATTACCAACATGGGC GCAGCACATAGTGATTTGGCATCC BUB3 CGGAACATGGGTTACGTGCAGC CCAAATACTCAACTGCCACTCGGC CAGE1 TCCAAAATGCACAGTCTTCTGGCT GGAGGCTCTTCAGTTTTTGCAGC CASP1 CCTGTTCCTGTGATGTGGAGGAAA GCTCTACCATCTGGCTGCTCAA CASP3 AGCGAATCAATGGACTCTGGAATATCC GTTTGCTGCATCGACATCTGTACCA CCL5 TCATTGCTACTGCCCTCTGCG ACTGCTGGGTTGGAGCACTTG CCL18 CCCTCCTTGTCCTCGTCTGCA GCACTGGGGGCTGGTTTCAG CCL26 TTCCAATACAGCCACAAGCCCC GGATGGGTACAGACTTTCTTGCCTC CCND2 TCAAGTGCGTGCAGAAGGACAT CTTCGCACTTCTGTTCCTCACA CCND3 TGGCTGCTGTGATTGCACATGA GATGGCGGGTACATGGCAAAGG CCR3 ACGCTGCTCTGCTTCCTGG TCCTCAGTTCCCCACCATCGC CCR4 AGCATCGTGCTTCCTGAGCAA GGTGTCTGCTATATCCGTGGGGT CCR7 AGACAGGGGTAGTGCGAGGC -

Molecular Update and Evolving Classification of Large B-Cell Lymphoma
cancers Review Molecular Update and Evolving Classification of Large B-Cell Lymphoma Arantza Onaindia 1,2,*, Nancy Santiago-Quispe 2, Erika Iglesias-Martinez 2 and Cristina Romero-Abrio 2 1 Bioaraba Health Research Institute, Oncohaematology Research Group, 01070 Vitoria-Gasteiz, Spain 2 Osakidetza Basque Health Service, Araba University Hospital, Pathology Department, 01070 Vitoria-Gasteiz, Spain; [email protected] (N.S.-Q.); [email protected] (E.I.-M.); [email protected] (C.R.-A.) * Correspondence: [email protected]; Tel.: +34-699-639-645 Simple Summary: The development of high-throughput technologies in recent years has increased our understanding of the molecular complexity of lymphomas, providing new insights into the pathogenesis of large B-cell neoplasms and identifying different molecular biomarkers with prog- nostic impact, that lead to the revision of the World Health Organization consensus classification of lymphomas. This review addresses the main histopathological and molecular features of large B-cells lymphomas, providing an overview of the main recent novelties introduced by the last update of the consensus classification. Abstract: Diffuse large B-cell lymphomas (DLBCLs) are aggressive B-cell neoplasms with consid- erable clinical, biologic, and pathologic diversity. The application of high throughput technologies to the study of lymphomas has yielded abundant molecular data leading to the identification of Citation: Onaindia, A.; distinct molecular identities and novel pathogenetic pathways. In light of this new information, Santiago-Quispe, N.; newly refined diagnostic criteria have been established in the fourth edition of the World Health Iglesias-Martinez, E.; Romero-Abrio, Organization (WHO) consensus classification of lymphomas, which was revised in 2016.