Identification of Potential Gene Signatures Associated with Osteosarcoma by Integrated Bioinformatics Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pancreatic Fibroblast Heterogeneity: from Development to Cancer
cells Review Pancreatic Fibroblast Heterogeneity: From Development to Cancer 1, 2, 2,3 Paloma E. Garcia y, Michael K. Scales y, Benjamin L. Allen and Marina Pasca di Magliano 2,3,4,* 1 Program in Molecular and Cellular Pathology, University of Michigan Medical School, University of Michigan, Ann Arbor, MI 48105, USA; [email protected] 2 Department of Cell and Developmental Biology, University of Michigan Medical School, University of Michigan, Ann Arbor, MI 48109, USA; [email protected] (M.K.S.); [email protected] (B.L.A.) 3 Rogel Cancer Center, Michigan Medicine, University of Michigan, Ann Arbor, MI 48109, USA 4 Department of Surgery, Michigan Medicine, University of Michigan, Ann Arbor, MI 48109, USA * Correspondence: [email protected]; Tel.: +1-734-276-7104 Authors contributed equally to this work. y Received: 22 October 2020; Accepted: 10 November 2020; Published: 12 November 2020 Abstract: Pancreatic ductal adenocarcinoma (PDA) is characterized by an extensive fibroinflammatory microenvironment that accumulates from the onset of disease progression. Cancer-associated fibroblasts (CAFs) are a prominent cellular component of the stroma, but their role during carcinogenesis remains controversial, with both tumor-supporting and tumor-restraining functions reported in different studies. One explanation for these contradictory findings is the heterogeneous nature of the fibroblast populations, and the different roles each subset might play in carcinogenesis. Here, we review the current literature on the origin and function of pancreatic fibroblasts, from the developing organ to the healthy adult pancreas, and throughout the initiation and progression of PDA. We also discuss clinical approaches to targeting fibroblasts in PDA. Keywords: fibroblasts; pancreas; cancer-associated fibroblasts; pancreas development; pancreatic cancer; tumor microenvironment; hedgehog; transforming growth factor Beta 1. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Cells Using Gene Expression Profiling and Insulin Gene Methylation
RESEARCH ARTICLE Comparison of enteroendocrine cells and pancreatic β-cells using gene expression profiling and insulin gene methylation ☯ ☯ Gyeong Ryul Ryu , Esder Lee , Jong Jin Kim, Sung-Dae MoonID, Seung-Hyun Ko, Yu- Bae Ahn, Ki-Ho SongID* Division of Endocrinology & Metabolism, Department of Internal Medicine, College of Medicine, The Catholic University of Korea, Seoul, Korea ☯ These authors contributed equally to this work. a1111111111 * [email protected] a1111111111 a1111111111 a1111111111 a1111111111 Abstract Various subtypes of enteroendocrine cells (EECs) are present in the gut epithelium. EECs and pancreatic β-cells share similar pathways of differentiation during embryonic develop- ment and after birth. In this study, similarities between EECs and β-cells were evaluated in OPEN ACCESS detail. To obtain specific subtypes of EECs, cell sorting by flow cytometry was conducted Citation: Ryu GR, Lee E, Kim JJ, Moon S-D, Ko S- from STC-1 cells (a heterogenous EEC line), and each single cell was cultured and pas- H, Ahn Y-B, et al. (2018) Comparison of saged. Five EEC subtypes were established according to hormone expression, measured enteroendocrine cells and pancreatic β-cells using by quantitative RT-PCR and immunostaining: L, K, I, G and S cells expressing glucagon-like gene expression profiling and insulin gene methylation. PLoS ONE 13(10): e0206401. https:// peptide-1, glucose-dependent insulinotropic polypeptide, cholecystokinin, gastrin and doi.org/10.1371/journal.pone.0206401 secretin, respectively. Each EEC subtype was found to express not only the corresponding Editor: Wataru Nishimura, International University gut hormone but also other gut hormones. Global microarray gene expression profiles of Health and Welfare School of Medicine, JAPAN revealed a higher similarity between each EEC subtype and MIN6 cells (a β-cell line) than Received: March 29, 2018 between C2C12 cells (a myoblast cell line) and MIN6 cells, and all EEC subtypes were highly similar to each other. -

Ten Commandments for a Good Scientist
Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Wageningen 2010 Thesis committee Thesis supervisors Prof. dr. ir. Ivonne M.C.M. Rietjens Professor of Toxicology Wageningen University Prof. dr. Albertinka J. Murk Personal chair at the sub-department of Toxicology Wageningen University Thesis co-supervisor Dr. ir. Jacques J.M. Vervoort Associate professor at the Laboratory of Biochemistry Wageningen University Other members Prof. dr. Michael R. Muller, Wageningen University Prof. dr. ir. Huub F.J. Savelkoul, Wageningen University Prof. dr. Everardus J. van Zoelen, Radboud University Nijmegen Dr. ir. Toine F.H. Bovee, RIKILT, Wageningen This research was conducted under the auspices of the Graduate School VLAG Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Thesis submitted in fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Tuesday 14 September 2010 at 4 p.m. in the Aula Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds. Ana María Sotoca Covaleda Thesis Wageningen University, Wageningen, The Netherlands, 2010, With references, and with summary in Dutch. ISBN: 978-90-8585-707-5 “Caminante no hay camino, se hace camino al andar. Al andar se hace camino, y al volver la vista atrás se ve la senda que nunca se ha de volver a pisar” - Antonio Machado – A mi madre. -

HOXB6 Homeo Box B6 HOXB5 Homeo Box B5 WNT5A Wingless-Type
5 6 6 5 . 4 2 1 1 1 2 4 6 4 3 2 9 9 7 0 5 7 5 8 6 4 0 8 2 3 1 8 3 7 1 0 0 4 0 2 5 0 8 7 5 4 1 1 0 3 6 0 4 8 3 7 4 7 6 9 6 7 1 5 0 8 1 4 1 1 7 1 0 0 4 2 0 8 1 1 1 2 5 3 5 0 7 2 6 9 1 2 1 8 3 5 2 9 8 0 6 0 9 5 1 9 9 2 1 1 6 0 2 3 0 3 6 9 1 6 5 5 7 1 1 2 1 1 7 5 4 6 6 4 1 1 2 8 4 7 1 6 2 7 7 5 4 3 2 4 3 6 9 4 1 7 1 3 4 1 2 1 3 1 1 4 7 3 1 1 1 1 5 3 2 6 1 5 1 3 5 4 5 2 3 1 1 6 1 7 3 2 5 4 3 1 6 1 5 3 1 7 6 5 1 1 1 4 6 1 6 2 7 2 1 2 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S HOXB6 homeo box B6 HOXB5 homeo box B5 WNT5A wingless-type MMTV integration site family, member 5A WNT5A wingless-type MMTV integration site family, member 5A FKBP11 FK506 binding protein 11, 19 kDa EPOR erythropoietin receptor SLC5A6 solute carrier family 5 sodium-dependent vitamin transporter, member 6 SLC5A6 solute carrier family 5 sodium-dependent vitamin transporter, member 6 RAD52 RAD52 homolog S. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Bioinformatic Analysis Reveals the Importance of Epithelial-Mesenchymal Transition in the Development of Endometriosis
www.nature.com/scientificreports OPEN Bioinformatic analysis reveals the importance of epithelial- mesenchymal transition in the development of endometriosis Meihong Chen1,6, Yilu Zhou2,3,6, Hong Xu4, Charlotte Hill2, Rob M. Ewing2,3, Deming He1, Xiaoling Zhang1 ✉ & Yihua Wang2,3,5 ✉ Background: Endometriosis is a frequently occurring disease in women, which seriously afects their quality of life. However, its etiology and pathogenesis are still unclear. Methods: To identify key genes/ pathways involved in the pathogenesis of endometriosis, we recruited 3 raw microarray datasets (GSE11691, GSE7305, and GSE12768) from Gene Expression Omnibus database (GEO), which contain endometriosis tissues and normal endometrial tissues. We then performed in-depth bioinformatic analysis to determine diferentially expressed genes (DEGs), followed by gene ontology (GO), Hallmark pathway enrichment and protein-protein interaction (PPI) network analysis. The fndings were further validated by immunohistochemistry (IHC) staining in endometrial tissues from endometriosis or control patients. Results: We identifed 186 DEGs, of which 118 were up-regulated and 68 were down-regulated. The most enriched DEGs in GO functional analysis were mainly associated with cell adhesion, infammatory response, and extracellular exosome. We found that epithelial-mesenchymal transition (EMT) ranked frst in the Hallmark pathway enrichment. EMT may potentially be induced by infammatory cytokines such as CXCL12. IHC confrmed the down-regulation of E-cadherin (CDH1) and up-regulation of CXCL12 in endometriosis tissues. Conclusions: Utilizing bioinformatics and patient samples, we provide evidence of EMT in endometriosis. Elucidating the role of EMT will improve the understanding of the molecular mechanisms involved in the development of endometriosis. Endometriosis is a frequently occurring gynaecological disease characterised by chronic pelvic pain, dysmenor- rhea and infertility1. -

Evidence for a Functional Role of Epigenetically Regulated Midcluster HOXB Genes in the Development of Barrett Esophagus
Evidence for a functional role of epigenetically regulated midcluster HOXB genes in the development of Barrett esophagus Massimiliano di Pietroa, Pierre Lao-Sirieixa, Shelagh Boyleb, Andy Cassidyc, Dani Castillod, Amel Saadic, Ragnhild Eskelandb,1, and Rebecca C. Fitzgeralda,2 aMedical Research Council Cancer Cell Unit, Hutchison Medical Research Council Research Centre, CB2 0XZ Cambridge, United Kingdom; bMedical Research Council Human Genetics Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, EH4 2XU Edinburgh, United Kingdom; cCancer Research United Kingdom, Cambridge Research Institute, Li Ka Shing Centre, CB2 0RE Cambridge, United Kingdom; and dSection of Gastrointestinal Surgery, Hospital Universitari del Mar, Universitat Autónoma de Barcelona, 08003 Barcelona, Spain Edited* by Walter Fred Bodmer, Weatherall Institute of Molecular Medicine, Oxford University, Oxford, United Kingdom, and approved April 18, 2012 (received for review October 17, 2011) Barrett esophagus (BE) is a human metaplastic condition that is the CDXs on their own are sufficient to induce an intestinal pheno- only known precursor to esophageal adenocarcinoma. BE is charac- type in the mouse esophagus. terized by a posterior intestinal-like phenotype in an anterior organ An intestinal-like epithelium in the esophagus is reminiscent of and therefore it is reminiscent of homeotic transformations, which homeotic transformations, which have been linked to mutations of can occur in transgenic animal models during embryonic develop- HOX genes (10). The 39 human HOX genes are divided into four ment as a consequence of mutations in HOX genes. In humans, clusters (HOXA, HOXB, HOXC, and HOXD) and have a collin- acquired deregulation of HOX genes during adulthood has been ear expression during development along the anterior-posterior linked to carcinogenesis; however, little is known about their role (A-P) axis, whereby 3′ end genes are activated earlier and are more in the pathogenesis of premalignant conditions. -

Genomic and Expression Profiling of Human Spermatocytic Seminomas: Primary Spermatocyte As Tumorigenic Precursor and DMRT1 As Candidate Chromosome 9 Gene
Research Article Genomic and Expression Profiling of Human Spermatocytic Seminomas: Primary Spermatocyte as Tumorigenic Precursor and DMRT1 as Candidate Chromosome 9 Gene Leendert H.J. Looijenga,1 Remko Hersmus,1 Ad J.M. Gillis,1 Rolph Pfundt,4 Hans J. Stoop,1 Ruud J.H.L.M. van Gurp,1 Joris Veltman,1 H. Berna Beverloo,2 Ellen van Drunen,2 Ad Geurts van Kessel,4 Renee Reijo Pera,5 Dominik T. Schneider,6 Brenda Summersgill,7 Janet Shipley,7 Alan McIntyre,7 Peter van der Spek,3 Eric Schoenmakers,4 and J. Wolter Oosterhuis1 1Department of Pathology, Josephine Nefkens Institute; Departments of 2Clinical Genetics and 3Bioinformatics, Erasmus Medical Center/ University Medical Center, Rotterdam, the Netherlands; 4Department of Human Genetics, Radboud University Medical Center, Nijmegen, the Netherlands; 5Howard Hughes Medical Institute, Whitehead Institute and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts; 6Clinic of Paediatric Oncology, Haematology and Immunology, Heinrich-Heine University, Du¨sseldorf, Germany; 7Molecular Cytogenetics, Section of Molecular Carcinogenesis, The Institute of Cancer Research, Sutton, Surrey, United Kingdom Abstract histochemistry, DMRT1 (a male-specific transcriptional regulator) was identified as a likely candidate gene for Spermatocytic seminomas are solid tumors found solely in the involvement in the development of spermatocytic seminomas. testis of predominantly elderly individuals. We investigated these tumors using a genome-wide analysis for structural and (Cancer Res 2006; 66(1): 290-302) numerical chromosomal changes through conventional kar- yotyping, spectral karyotyping, and array comparative Introduction genomic hybridization using a 32 K genomic tiling-path Spermatocytic seminomas are benign testicular tumors that resolution BAC platform (confirmed by in situ hybridization). -

The Homeobox Gene CDX2 Is Aberrantly Expressed and Associated with an Inferior Prognosis in Patients with Acute Lymphoblastic Leukemia
Leukemia (2009) 23, 649–655 & 2009 Macmillan Publishers Limited All rights reserved 0887-6924/09 $32.00 www.nature.com/leu ORIGINAL ARTICLE The homeobox gene CDX2 is aberrantly expressed and associated with an inferior prognosis in patients with acute lymphoblastic leukemia S Thoene1,2,4, VPS Rawat1,2,4, B Heilmeier1, E Hoster1, KH Metzeler1, T Herold1, W Hiddemann1,2,NGo¨kbuget3, D Hoelzer3, SK Bohlander1,2, M Feuring-Buske1,2 and C Buske1,2 1Department of Medicine III, Klinikum Grosshadern, Munich, Germany; 2Clinical Cooperative Group Leukemia, Helmholtz Center Munich for Environmental Health, Munich, Germany and 3Department of Medicine III, Johann Wolfgang Goethe University, Frankfurt/Main, Germany Molecular characterization of acute lymphoblastic leukemia gene deregulation.11 However, it was shown that besides Hox (ALL) has greatly improved the ability to categorize and genes Cdx2 regulates other stem cell regulatory genes, such as Scl, prognostify patients with this disease. In this study, we show 12 that the proto-oncogene CDX2 is aberrantly expressed in the Gata1 and Runx1. Thus, aberrant expression of CDX2 might majority of cases with B-lineage ALL and T-ALL. High expres- perturb the stem cell regulatory network at different levels. sion of CDX2 correlated significantly with the ALL subtype We now report that CDX2 is aberrantly expressed in 81% of pro-B ALL, cALL, Ph þ ALL and early T-ALL. Furthermore, high adult patients with ALL, and that high expression levels of this expression of CDX2 was associated with inferior overall proto-oncogene predict poor treatment outcome in patients with survival and showed up as a novel and strong risk factor for this disease. -

(Mouse) Ezh2 Blocking Peptide (Center) Synthetic Peptide Catalog # Bp21367c
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 (Mouse) Ezh2 Blocking Peptide (Center) Synthetic peptide Catalog # BP21367c Specification (Mouse) Ezh2 Blocking Peptide (Center) - (Mouse) Ezh2 Blocking Peptide (Center) - Product Background Information Polycomb group (PcG) protein. Catalytic Primary Accession Q61188 subunit of the PRC2/EED-EZH2 complex, which methylates (H3K9me) and 'Lys-27' (H3K27me) of histone H3, leading to transcriptional (Mouse) Ezh2 Blocking Peptide (Center) - Additional Information repression of the affected target gene. Able to mono-, di- and trimethylate 'Lys-27' of histone H3 to form H3K27me1, H3K27me2 and Gene ID 14056 H3K27me3, respectively. Compared to EZH2-containing complexes, it is more Other Names abundant in embryonic stem cells and plays a Histone-lysine N-methyltransferase EZH2, major role in forming H3K27me3, which is ENX-1, Enhancer of zeste homolog 2, Ezh2, required for embryonic stem cell identity and Enx1h proper differentiation. The PRC2/EED-EZH2 Target/Specificity complex may also serve as a recruiting The synthetic peptide sequence is selected platform for DNA methyltransferases, thereby from aa 260-275 of HUMAN Ezh2 linking two epigenetic repression systems. Genes repressed by the PRC2/EED-EZH2 Format complex include HOXA7, HOXB6 and HOXC8. Peptides are lyophilized in a solid powder EZH2 can also methylate non-histone proteins format. Peptides can be reconstituted in such as the transcription factor GATA4 and the solution using the appropriate buffer as nuclear receptor RORA. Regulates the needed. circadian clock via histone methylation at the promoter of the circadian genes. Essential for Storage the CRY1/2-mediated repression of the Maintain refrigerated at 2-8°C for up to 6 transcriptional activation of PER1/2 by the months. -
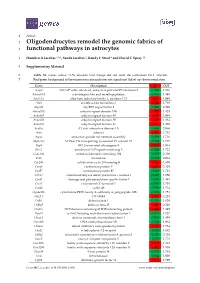
Oligodendrocytes Remodel the Genomic Fabrics of Functional Pathways in Astrocytes
1 Article 2 Oligodendrocytes remodel the genomic fabrics of 3 functional pathways in astrocytes 4 Dumitru A Iacobas 1,2,*, Sanda Iacobas 3, Randy F Stout 4 and David C Spray 2,5 5 Supplementary Material 6 Table S1. Genes whose >1.5x absolute fold-change did not meet the individual CUT criterion. 7 Red/green background of the expression ratio indicates not significant (false) up-/down-regulation. Gene Description X CUT Acap2 ArfGAP with coiled-coil, ankyrin repeat and PH domains 2 -1.540 1.816 Adamts18 a disintegrin-like and metallopeptidase -1.514 1.594 Akr1c12 aldo-keto reductase family 1, member C12 1.866 1.994 Alx3 aristaless-like homeobox 3 1.536 1.769 Alyref2 Aly/REF export factor 2 -1.880 2.208 Ankrd33b ankyrin repeat domain 33B 1.593 1.829 Ankrd45 ankyrin repeat domain 45 1.514 1.984 Ankrd50 ankyrin repeat domain 50 1.628 1.832 Ankrd61 ankyrin repeat domain 61 1.645 1.802 Arid1a AT rich interactive domain 1A -1.668 2.066 Artn artemin 1.524 1.732 Aspm abnormal spindle microtubule assembly -1.693 1.716 Atp6v1e1 ATPase, H+ transporting, lysosomal V1 subunit E1 -1.679 1.777 Bag4 BCL2-associated athanogene 4 1.723 1.914 Birc3 baculoviral IAP repeat-containing 3 -1.588 1.722 Ccdc104 coiled-coil domain containing 104 -1.819 2.130 Ccl2 chemokine -1.699 2.034 Cdc20b cell division cycle 20 homolog B 1.512 1.605 Cenpf centromere protein F 2.041 2.128 Cep97 centrosomal protein 97 -1.641 1.723 COX1 mitochondrially encoded cytochrome c oxidase I -1.607 1.650 Cpsf7 cleavage and polyadenylation specific factor 7 -1.635 1.891 Crct1 cysteine-rich