FINAL TECHNICAL REPORT NEIL W. ASHCROFT PROFESSOR of PHYSICS August 1972 To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

197 3Apj. . .186.1107S the Astrophysical Journal, 186:1107
The Astrophysical Journal, 186:1107-1125, 1973 December 15 .186.1107S . © 1973. The American Astronomical Society. All rights reserved. Printed in U.S.A. 3ApJ. 197 PHASE EQUILIBRIA IN MOLECULAR HYDROGEN-HELIUM MIXTURES AT HIGH PRESSURES* W. B. Streett Science Research Laboratory, U.S. Military Academy, West Point, New York Received 1973 June 11 ABSTRACT Experiments on phase behavior in hydrogen-helium mixtures have been carried out at pressures up to 9.3 kilobars, at temperatures from 26° to 100° K. Two distinct fluid phases are shown to exist at supercritical temperatures and high pressures. Both the trend of the experimental results and an analysis based on the van der Waals theory of mixtures suggest that this fluid-fluid phase separation persists at temperatures and pressures beyond the range of these experiments, perhaps even to the limits of stability of the molecular phases. The results confirm earlier predictions concerning the form of the hydrogen-helium phase diagram in the region of pressure-induced solidification of the molecular phases at supercritical temperatures. The implications of this phase diagram for planetary interiors are discussed. Subject headings: gas dynamics — interiors, planetary — molecules I. INTRODUCTION The properties of hydrogen-helium mixtures are of interest for several reasons. From a theoretical standpoint, they are of interest because they are composed of the two elements with the simplest atomic and molecular structures, and would seem to be among the mixtures most amenable to a theoretical treatment based on first principles. However, it is fair to say that a satisfactory theory of dense mixtures of molecular hydrogen and helium has yet to be developed. -

Effects of Helium Phase Separation on the Evolution of Extrasolar Giant Planets
Accepted to ApJ, February, 2004 A Preprint typeset using L TEX style emulateapj v. 11/12/01 EFFECTS OF HELIUM PHASE SEPARATION ON THE EVOLUTION OF EXTRASOLAR GIANT PLANETS Jonathan J. Fortney1, W. B. Hubbard1 Accepted to ApJ, February, 2004 ABSTRACT We build on recent new evolutionary models of Jupiter and Saturn and here extend our calculations to investigate the evolution of extrasolar giant planets of mass 0.15 to 3.0 MJ. Our inhomogeneous thermal history models show that the possible phase separation of helium from liquid metallic hydrogen in the deep interiors of these planets can lead to luminosities ∼ 2 times greater than have been predicted by homogeneous models. For our chosen phase diagram this phase separation will begin to affect the planets’ evolution at ∼ 700 Myr for a 0.15 MJ object and ∼ 10 Gyr for a 3.0 MJ object. We show how phase separation affects the luminosity, effective temperature, radii, and atmospheric helium mass fraction as a function of age for planets of various masses, with and without heavy element cores, and with and without the effect of modest stellar irradiation. This phase separation process will likely not affect giant planets within a few AU of their parent star, as these planets will cool to their equilibrium temperatures, determined by stellar heating, before the onset of phase separation. We discuss the detectability of these objects and the likelihood that the energy provided by helium phase separation can change the timescales for formation and settling of ammonia clouds by several Gyr. We discuss how correctly incorporating stellar irradiation into giant planet atmosphere and albedo modeling may lead to a consistent evolutionary history for Jupiter and Saturn. -

Part 1: the 1.7 and 3.9 Earth Radii Rule
Hi this is Steve Nerlich from Cheap Astronomy www.cheapastro.com and this is What are exoplanets made of? Part 1: The 1.7 and 3.9 earth Radii rule. As of August 2016, the current count of confirmed exoplanets is up around 3,400 in 2,617 systems – with 590 of those systems confirmed to be multiplanet systems. And the latest thinking is that if you want to understand what exoplanets are made of you need to appreciate the physical limits of planet-hood, which are defined by the boundaries of 1.7 and 3.9 Earth radii . Consider that the make-up of an exoplanet is largely determined by the elemental make up of its protoplanetary disk. While most material in the Universe is hydrogen and helium – these are both tenuous gases. In order to generate enough gravity to hang on to them, you need a lot of mass to start with. So, if you’re Earth, or anything up to 1.7 times the radius of Earth – you’ve got no hope of hanging onto more than a few traces of elemental hydrogen and helium. Indeed, any exoplanet that’s less than 1.7 Earth radii has to be primarily composed of non-volatiles – that is, things that don’t evaporate or blow away easily – to have any chance of gravitationally holding together. A non-volatile exoplanet might be made of rock – which for our Solar System is a primarily silicon/oxygen based mineral matrix, but as we’ll hear, small sub-1.7 Earth radii exoplanets could be made of a whole range of other non-volatile materials. -

Shallow Ultraviolet Transits of WD 1145+017
Shallow Ultraviolet Transits of WD 1145+017 Item Type Article Authors Xu, Siyi; Hallakoun, Na’ama; Gary, Bruce; Dalba, Paul A.; Debes, John; Dufour, Patrick; Fortin-Archambault, Maude; Fukui, Akihiko; Jura, Michael A.; Klein, Beth; Kusakabe, Nobuhiko; Muirhead, Philip S.; Narita, Norio; Steele, Amy; Su, Kate Y. L.; Vanderburg, Andrew; Watanabe, Noriharu; Zhan, Zhuchang; Zuckerman, Ben Citation Siyi Xu et al 2019 AJ 157 255 DOI 10.3847/1538-3881/ab1b36 Publisher IOP PUBLISHING LTD Journal ASTRONOMICAL JOURNAL Rights Copyright © 2019. The American Astronomical Society. All rights reserved. Download date 09/10/2021 04:17:12 Item License http://rightsstatements.org/vocab/InC/1.0/ Version Final published version Link to Item http://hdl.handle.net/10150/634682 The Astronomical Journal, 157:255 (12pp), 2019 June https://doi.org/10.3847/1538-3881/ab1b36 © 2019. The American Astronomical Society. All rights reserved. Shallow Ultraviolet Transits of WD 1145+017 Siyi Xu (许偲艺)1 ,Na’ama Hallakoun2 , Bruce Gary3 , Paul A. Dalba4 , John Debes5 , Patrick Dufour6, Maude Fortin-Archambault6, Akihiko Fukui7,8 , Michael A. Jura9,21, Beth Klein9 , Nobuhiko Kusakabe10, Philip S. Muirhead11 , Norio Narita (成田憲保)8,10,12,13,14 , Amy Steele15, Kate Y. L. Su16 , Andrew Vanderburg17 , Noriharu Watanabe18,19, Zhuchang Zhan (詹筑畅)20 , and Ben Zuckerman9 1 Gemini Observatory, 670 N. A’ohoku Place, Hilo, HI 96720, USA; [email protected] 2 School of Physics and Astronomy, Tel-Aviv University, Tel-Aviv 6997801, Israel 3 Hereford Arizona Observatory, Hereford, AZ 85615, USA -

NASA Data Used to Discover Eighth Planet Circling Distant Star
Journal of Aircraft and Spacecraft Technology Review NASA Data Used to Discover Eighth Planet Circling Distant Star 1 2 2 1 Relly Victoria Petrescu, Raffaella Aversa, Antonio Apicella and Florian Ion Tiberiu Petrescu 1ARoTMM-IFToMM, Bucharest Polytechnic University, Bucharest, (CE), Romania 2Department of Architecture and Industrial Design, Advanced Material Lab, Second University of Naples, 81031 Aversa (CE), Italy Article history Abstract: Discovering the origins of life and colonizing extraterrestrial planets Received: 09-01-2018 are two of the greatest ambitions that the international scientific community has Revised: 13-01-2018 today. Today, NASA is more prepared than ever to embark on an Accepted: 18-01-2018 unprecedented race to conquer other worlds. The Kepler mission, named after Johannes Kepler, the German astronomer of the 16th-17th centuries, was Corresponding Author: Florian Ion Tiberiu Petrescu scheduled to begin on March 7, 2009. In this mission, Space Agency released ARoTMM-IFToMM, space from the Cape Canaveral Air Force Base, Florida, a Delta II rocket that Bucharest Polytechnic will carry the Kepler space probe. It will try to identify Earth-like planets that University, Bucharest, (CE), orbitate stars similar to the Sun in a warm area of space, with liquid water and Romania oxygen, that is, heavenly bodies that have all the conditions of life formation E-mail: [email protected] and maintenance. Kepler is looking for a new Earth. Kepler is a critical component of NASA's efforts to identify and study planets with Earth-like eco- conditions. The results of the Kepler space photometer will prove to be extremely important in understanding the frequency of Earth-sized planets in our galaxy. -

Analysis of Exoplanetary Transit Light Curves Joshua Adam Carter
Analysis of Exoplanetary Transit Light Curves by Joshua Adam Carter B.S., Physics, University of North Carolina at Chapel Hill, 2004 B.S., Mathematics, University of North Carolina at Chapel Hill, 2004 Submitted to the Department of Physics in partial fulfillment of the requirements for the degree oft MASSACHUSETTS INSTITUTE Doctor of Philosophy at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY ARCHIVES September 2009 © Massachusetts Institute of Technology 2009. All rights reserved. Author ................ .............. Department of Physics 31, 2009 7A .August Certified by....... Joshua N. Winn Assistant Professor of Physics Class of 1942 Career Development Professor Thesis Supervisor A Accepted by ............................ TlW rs J. Greytak Lester Wolfe Professor of Physics Associate Department Head for Education Analysis of Exoplanetary Transit Light Curves by Joshua Adam Carter Submitted to the Department of Physics on August 31, 2009, in partial fulfillment of the requirements for the degree of Doctor of Philosophy Abstract This Thesis considers the scenario in which an extra-solar planet (exoplanet) passes in front of its star relative to our observing perspective. In this event, the light curve measured for the host star features a systematic drop in flux occurring once every orbital period as the exoplanet covers a portion of the stellar disk. This exoplanetary transit light curve provides a wealth of information about both the planet and star. In this Thesis we consider the transit light curve as a tool for characterizing the exoplanet. The Thesis can divided into two parts. In the first part, comprised of the second and third chapters, I assess what ob- servables describing the exoplanet (and host) may be measured, how well they can be measured, and what effect systematics in the light curve can have on our estimation of these parameters. -

Lecture 19 Giant Planets I Great Red Spot
Lecture 19 Giant Planets I Great Red Spot Jupiter The largest planet in the solar system Mass = 300 x Earth Mass = 0.001 x Sun Atmospheric Compositions Jupiter Extensive Ring System Mass = 95 x Earth = 1/3 Jupiter Rings can appear to disappear when we see them edge on Uranus is Uranus blue but very bland Mass = 14.5 x Earth = 1/20 Jupiter Lecture 21 Uranus and Neptune Mass = 17 x Earth This is a = 1/18 Jupiter Voyager 2 image of Neptune’s Neptune Great Dark Spot - Blue has now disappeared We will discuss and compare the four Jovian planets together Orbital and Physical Properties Internal Structures Atmospheres Magnetospheres Satellites Rings !"##"$%#&'()*('+"%$),)-".&"/) Small!"##"$%#&'()*('+"%$),)-".&"/) and rocky - no rings - few satellites Mercury Venus Earth Mars (radar) Jovian Planets Jovian Planets :';$<*/*&+'=$&4'">'()%&*+':';$<*/*&+'=$&4'">'()%&*+' •! !"#$%&'()%&*+,-'".+*/' •! !"#$%&'()%&*+,-'".+*/' ,")%/',0,+*12'3*0"&4' ,")%/',0,+*12'3*0"&4' 5%/,' 5%/,' –!!.6$+*/' –!!.6$+*/' –!7%+./&' –!7%+./&' –!8/%&.,' –!8/%&.,' •–!9*6+.&*' –!9*6+.&*'! !"##$%&'&()%$&(*++",$& •! -).$%&/$0+"123&/"4$%$01&5)(6)+"7)0& •! 8"0#+& •! 9:($%):+&())0+& Knowledge is mostly very recent and derived from US space missions Space Missions To Outer Planets Voyager Spacecraft used the gravity of the planets to reach the outer solar system - a planetary sling-shot !"#$%&'()(*(+( Outer planet arrangement ~ (1970s-1980s) every 175 years! 4 giant planets: Jupiter Saturn Uranus Neptune Orbit the Sun in the outer solar system - beyond the “snow” line - outside the Asteroid Belt and inside the Kuiper Belt !"#$%&#'()*+,-+./0+$.) 1%,-%'#&2'%)3#'/%.)4/&5)6/.&#$7%)&+)82$)#$6)'%92"#&%.:) •! ;5/75)%"%,%$&.)#7&2#""()7+$6%$.%) •! ;5/75)7+,-+2$6.)#'%)<+',%6)<'+,)&5%)%"%,%$&.) •! =&)45#&)'#&%.)&5%)7+,-+2$6.)#'%)<+',%6>) Volatile species will only be stable beyond a “snow line”. -

Bibliography from ADS File: Chaplin.Bib May 31, 2021 1
Bibliography from ADS file: chaplin.bib Metcalfe, T. S., van Saders, J. L., Basu, S., et al., “The Evolution of Rota- August 16, 2021 tion and Magnetic Activity in 94 Aqr Aa from Asteroseismology with TESS”, 2020ApJ...900..154M ADS Nielsen, M. B., Ball, W. H., Standing, M. R., et al., “TESS asteroseismology of Zinn, J. C., Stello, D., Elsworth, Y., et al., “The K2 Galactic Archaeology the known planet host star λ2 Fornacis”, 2020A&A...641A..25N ADS Program Data Release 3: Age-abundance patterns in C1-C8, C10-C18”, Steinmetz, M., Guiglion, G., McMillan, P. J., et al., “The Sixth Data Release of 2021arXiv210805455Z ADS the Radial Velocity Experiment (RAVE). II. Stellar Atmospheric Parameters, Lyttle, A. J., Davies, G. R., Li, T., et al., “Hierarchically modelling Kepler Chemical Abundances, and Distances”, 2020AJ....160...83S ADS dwarfs and subgiants to improve inference of stellar properties with astero- Steinmetz, M., Matijevic,ˇ G., Enke, H., et al., “The Sixth Data Release of the Ra- seismology”, 2021MNRAS.505.2427L ADS dial Velocity Experiment (RAVE). I. Survey Description, Spectra, and Radial Hill, M. L., Kane, S. R., Campante, T. L., et al., “Asteroseismology of Velocities”, 2020AJ....160...82S ADS iota Draconis and Discovery of an Additional Long-Period Companion”, Ahumada, R., Prieto, C. A., Almeida, A., et al., “The 16th Data Release of 2021arXiv210713583H ADS the Sloan Digital Sky Surveys: First Release from the APOGEE-2 Southern Thomas, A. E. L., Chaplin, W. J., Basu, S., et al., “Impact of magnetic Survey and Full Release of eBOSS Spectra”, 2020ApJS..249....3A ADS activity on inferred stellar properties of main-sequence Sun-like stars”, Jiang, C., Bedding, T. -

Planets Solar System Paper Contents
Planets Solar system paper Contents 1 Jupiter 1 1.1 Structure ............................................... 1 1.1.1 Composition ......................................... 1 1.1.2 Mass and size ......................................... 2 1.1.3 Internal structure ....................................... 2 1.2 Atmosphere .............................................. 3 1.2.1 Cloud layers ......................................... 3 1.2.2 Great Red Spot and other vortices .............................. 4 1.3 Planetary rings ............................................ 4 1.4 Magnetosphere ............................................ 5 1.5 Orbit and rotation ........................................... 5 1.6 Observation .............................................. 6 1.7 Research and exploration ....................................... 6 1.7.1 Pre-telescopic research .................................... 6 1.7.2 Ground-based telescope research ............................... 7 1.7.3 Radiotelescope research ................................... 8 1.7.4 Exploration with space probes ................................ 8 1.8 Moons ................................................. 9 1.8.1 Galilean moons ........................................ 10 1.8.2 Classification of moons .................................... 10 1.9 Interaction with the Solar System ................................... 10 1.9.1 Impacts ............................................ 11 1.10 Possibility of life ........................................... 12 1.11 Mythology ............................................. -

Transits and Occultations
Transits and Occultations Joshua N. Winn Massachusetts Institute of Technology When we are fortunate enough to view an exoplanetary system nearly edge-on, the star and planet periodically eclipse each other. Observations of eclipses—transits and occultations—provide a bonanza of information that cannot be obtained from radial-velocity data alone, such as the relative dimensions of the planet and its host star, as well as the orientation of the planet’s orbit relative to the sky plane and relative to the stellar rotation axis. The wavelength-dependence of the eclipse signal gives clues about the the temperature and composition of the planetary atmosphere. Anomalies in the timing or other properties of the eclipses may betray the presence of additional planets or moons. Searching for eclipses is also a productive means of discovering new planets. This chapter reviews the basic geometry and physics of eclipses, and summarizes the knowledge that has been gained through eclipse observations, as well as the information that might be gained in the future. 1. INTRODUCTION and the passage of the smaller body behind the larger body is an occultation. Formally, transits are cases when the full From immemorial antiquity, men have dreamed of a disk of the smaller body passes completely within that of royal road to success—leading directly and easily to some the larger body, and occultations refer to the complete con- goal that could be reached otherwise only by long ap- cealment of the smaller body. We will allow those terms to proaches and with weary toil. Times beyond number, this include the grazing cases in which the bodies’ silhouettes dream has proved to be a delusion... -

Transiting Exoplanets from the Corot Space Mission II
A&A 482, L21–L24 (2008) Astronomy DOI: 10.1051/0004-6361:200809431 & c ESO 2008 Astrophysics Letter to the Editor Transiting exoplanets from the CoRoT space mission II. CoRoT-Exo-2b: a transiting planet around an active G star, R. Alonso1, M. Auvergne2,A.Baglin2, M. Ollivier3, C. Moutou1, D. Rouan2,H.J.Deeg4, S. Aigrain5, J. M. Almenara4, M. Barbieri1,P.Barge1,W.Benz6, P. Bordé3, F. Bouchy7,R.DelaReza8,M.Deleuil1,R.Dvorak9, A. Erikson10,M.Fridlund11, M. Gillon12, P. Gondoin11, T. Guillot13, A. Hatzes14, G. Hébrard7, P. Kabath10,L.Jorda1, H. Lammer15, A. Léger3, A. Llebaria1, B. Loeillet1,7, P. Magain16, M. Mayor12, T. Mazeh17,M.Pätzold18,F.Pepe12, F. Pont12,D.Queloz12,H.Rauer10,19, A. Shporer17, J. Schneider20, B. Stecklum14,S.Udry12, and G. Wuchterl14 (Affiliations can be found after the references) Received 21 January 2008 / Accepted 27 February 2008 ABSTRACT Context. The CoRoT mission, a pioneer in exoplanet searches from space, has completed its first 150 days of continuous observations of ∼12 000 stars in the galactic plane. An analysis of the raw data identifies the most promising candidates and triggers the ground-based follow-up. Aims. We report on the discovery of the transiting planet CoRoT-Exo-2b, with a period of 1.743 days, and characterize its main parameters. Methods. We filter the CoRoT raw light curve of cosmic impacts, orbital residuals, and low frequency signals from the star. The folded light curve of 78 transits is fitted to a model to obtain the main parameters. Radial velocity data obtained with the SOPHIE, CORALIE and HARPS spectro- graphs are combined to characterize the system. -
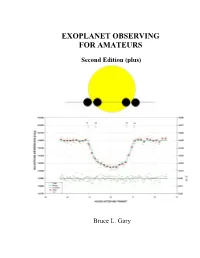
Book Describing Techniques to Detect Transiting Exoplanets
EXOPLANET OBSERVING FOR AMATEURS Second Edition (plus) Bruce L. Gary Books by Bruce L. Gary ESSAYS FROM ANOTHER PARADIGM, 1992, 1993 (Abridged Edition) GENETIC ENSLAVEMENT: A CALL TO ARMS FOR INDIVIDUAL LIBERATION, 2004, 2006, 2008 THE MAKING OF A MISANTHROPE: BOOK 1, AN AUTOBIOGRAPHY, 2005 A MISANTHROPE’S HOLIDAY: VIGNETTES AND STORIES, 2007 EXOPLANET OBSERVING FOR AMATEURS (First Edition), 2007 QUOTES FOR MISANTHROPES: MOCKING HOMO HYPOCRITUS, 2007 ii EXOPLANET OBSERVING FOR AMATEURS THIRD EDITION Bruce L. Gary Reductionist Publications, d/b/a 5320 E. Calle Manzana Hereford, AZ 85615 iii Bruce L. Gary 5320 E. Calle Manzana Hereford, AZ 85615; SA [email protected] Cover Illustration: WASP-10 light curve, 2008 November 14, by the author, using a 14-inch Meade LX200 telescope and an SBIG ST-8XE CCD, unfiltered. The sketch is a scale- accurate depiction of exoplanet WASP-10b transiting in front of its parent star, WASP-10a. Copyright 2010 by Bruce L. Gary All rights reserved except for brief passages quoted in a review. No part of this book may be reproduced, stored in a retrieval system, or transmitted in any form and by any means: electronic, mechanical, photocopying, recording or otherwise without express prior permission from the publisher. Requests for usage permission or additional information should be addressed to: “Bruce L. Gary” <[email protected]> or Reductionist Publications, d/b/a 5320 E. Calle Manzana Hereford, AZ 85615 Second Edition (plus): 2010 October 22 (typo and other corrections) Second Edition: 2009 November 10 First edition: 2007 August Printed by Mira Digital Publishing, St. Louis, MO ISBN 978-0-9798446-3-8 iv v Dedicated to the memory of Carl Sagan A giant among men, who would have loved the excitement of exoplanet discoveries, which would have further inspired him to speculate about life in the universe.