Transcriptome Profiling of Arabian Horse Blood During Training Regimens
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Modulating Hallmarks of Cholangiocarcinoma
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Fall 12-14-2018 Modulating Hallmarks of Cholangiocarcinoma Cody Wehrkamp University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Molecular Biology Commons Recommended Citation Wehrkamp, Cody, "Modulating Hallmarks of Cholangiocarcinoma" (2018). Theses & Dissertations. 337. https://digitalcommons.unmc.edu/etd/337 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. MODULATING HALLMARKS OF CHOLANGIOCARCINOMA by Cody J. Wehrkamp A DISSERTATION Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor Justin L. Mott University of Nebraska Medical Center Omaha, Nebraska November 2018 Supervisory Committee: Kaustubh Datta, Ph.D. Melissa Teoh‐Fitzgerald, Ph.D. Richard G. MacDonald, Ph.D. Acknowledgements This endeavor has led to scientific as well as personal growth for me. I am indebted to many for their knowledge, influence, and support along the way. To my mentor, Dr. Justin L. Mott, you have been an incomparable teacher and invaluable guide. You upheld for me the concept that science is intrepid, even when the experience is trying. Through my training, and now here at the end, I can say that it has been an honor to be your protégé. When you have shaped your future graduates to be and do great, I will be privileged to say that I was your first one. -

Serum Albumin OS=Homo Sapiens
Protein Name Cluster of Glial fibrillary acidic protein OS=Homo sapiens GN=GFAP PE=1 SV=1 (P14136) Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 Cluster of Isoform 3 of Plectin OS=Homo sapiens GN=PLEC (Q15149-3) Cluster of Hemoglobin subunit beta OS=Homo sapiens GN=HBB PE=1 SV=2 (P68871) Vimentin OS=Homo sapiens GN=VIM PE=1 SV=4 Cluster of Tubulin beta-3 chain OS=Homo sapiens GN=TUBB3 PE=1 SV=2 (Q13509) Cluster of Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 (P60709) Cluster of Tubulin alpha-1B chain OS=Homo sapiens GN=TUBA1B PE=1 SV=1 (P68363) Cluster of Isoform 2 of Spectrin alpha chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTAN1 (Q13813-2) Hemoglobin subunit alpha OS=Homo sapiens GN=HBA1 PE=1 SV=2 Cluster of Spectrin beta chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTBN1 PE=1 SV=2 (Q01082) Cluster of Pyruvate kinase isozymes M1/M2 OS=Homo sapiens GN=PKM PE=1 SV=4 (P14618) Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 Clathrin heavy chain 1 OS=Homo sapiens GN=CLTC PE=1 SV=5 Filamin-A OS=Homo sapiens GN=FLNA PE=1 SV=4 Cytoplasmic dynein 1 heavy chain 1 OS=Homo sapiens GN=DYNC1H1 PE=1 SV=5 Cluster of ATPase, Na+/K+ transporting, alpha 2 (+) polypeptide OS=Homo sapiens GN=ATP1A2 PE=3 SV=1 (B1AKY9) Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 Dihydropyrimidinase-related protein 2 OS=Homo sapiens GN=DPYSL2 PE=1 SV=1 Cluster of Alpha-actinin-1 OS=Homo sapiens GN=ACTN1 PE=1 SV=2 (P12814) 60 kDa heat shock protein, mitochondrial OS=Homo -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Quantitative Live Cell Imaging Reveals Influenza Virus Manipulation Of
ARTICLE https://doi.org/10.1038/s41467-019-13838-3 OPEN Quantitative live cell imaging reveals influenza virus manipulation of Rab11A transport through reduced dynein association Amar R. Bhagwat 1, Valerie Le Sage1, Eric Nturibi1, Katarzyna Kulej2, Jennifer Jones 1, Min Guo3, Eui Tae Kim 2, Benjamin A. Garcia4,5, Matthew D. Weitzman2,5,6, Hari Shroff3 & Seema S. Lakdawala 1,7* fl 1234567890():,; Assembly of infectious in uenza A viruses (IAV) is a complex process involving transport from the nucleus to the plasma membrane. Rab11A-containing recycling endosomes have been identified as a platform for intracellular transport of viral RNA (vRNA). Here, using high spatiotemporal resolution light-sheet microscopy (~1.4 volumes/second, 330 nm isotropic resolution), we quantify Rab11A and vRNA movement in live cells during IAV infection and report that IAV infection decreases speed and increases arrest of Rab11A. Unexpectedly, infection with respiratory syncytial virus alters Rab11A motion in a manner opposite to IAV, suggesting that Rab11A is a common host component that is differentially manipulated by respiratory RNA viruses. Using two-color imaging we demonstrate co-transport of Rab11A and IAV vRNA in infected cells and provide direct evidence that vRNA-associated Rab11A have altered transport. The mechanism of altered Rab11A movement is likely related to a decrease in dynein motors bound to Rab11A vesicles during IAV infection. 1 Department of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, 450 Technology Drive, Pittsburgh, PA 15219, USA. 2 The Children’s Hospital of Philadelphia Research Institute, 3501 Civic Center Dr., Philadelphia, PA 19104, USA. 3 Section on High Resolution Optical Imaging, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 13 South Drive, Building 13, Bethesda, MD 20892, USA. -

NATURAL KILLER CELLS, HYPOXIA, and EPIGENETIC REGULATION of HEMOCHORIAL PLACENTATION by Damayanti Chakraborty Submitted to the G
NATURAL KILLER CELLS, HYPOXIA, AND EPIGENETIC REGULATION OF HEMOCHORIAL PLACENTATION BY Damayanti Chakraborty Submitted to the graduate degree program in Pathology and Laboratory Medicine and the Graduate Faculty of the University of Kansas in partial fulfillment ofthe requirements for the degree of Doctor of Philosophy. ________________________________ Chair: Michael J. Soares, Ph.D. ________________________________ Jay Vivian, Ph.D. ________________________________ Patrick Fields, Ph.D. ________________________________ Soumen Paul, Ph.D. ________________________________ Michael Wolfe, Ph.D. ________________________________ Adam J. Krieg, Ph.D. Date Defended: 04/01/2013 The Dissertation Committee for Damayanti Chakraborty certifies that this is the approved version of the following dissertation: NATURAL KILLER CELLS, HYPOXIA, AND EPIGENETIC REGULATION OF HEMOCHORIAL PLACENTATION ________________________________ Chair: Michael J. Soares, Ph.D. Date approved: 04/01/2013 ii ABSTRACT During the establishment of pregnancy, uterine stromal cells differentiate into decidual cells and recruit natural killer (NK) cells. These NK cells are characterized by low cytotoxicity and distinct cytokine production. In rodent as well as in human pregnancy, the uterine NK cells peak in number around mid-gestation after which they decline. NK cells associate with uterine spiral arteries and are implicated in pregnancy associated vascular remodeling processes and potentially in modulating trophoblast invasion. Failure of trophoblast invasion and vascular remodeling has been shown to be associated with pathological conditions like preeclampsia syndrome, hypertension in mother and/or fetal growth restriction. We hypothesize that NK cells fundamentally contribute to the organization of the placentation site. In order to study the in vivo role of NK cells during pregnancy, gestation stage- specific NK cell depletion was performed in rats using anti asialo GM1 antibodies. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
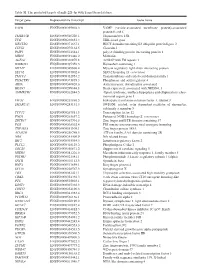
Table SI. the Predicted Targets of Mir-221-3P with Targetscan Database
Table SI. The predicted targets of miR-221-3p with TargetScan database. Target gene Representative transcript Gene name VAPB ENST00000395802.3 VAMP (vesicle-associated membrane protein)-associated protein B and C TMSB15B ENST00000540220.1 Thymosin beta 15B TFG ENST00000240851.4 TRK-fused gene HECTD2 ENST00000371667.1 HECT domain containing E3 ubiquitin protein ligase 2 CLVS2 ENST00000275162.5 Clavesin 2 PAIP1 ENST00000514514.1 poly(A) binding protein interacting protein 1 MIDN ENST00000591446.2 Midnolin AGFG1 ENST00000310078.8 ArfGAP with FG repeats 1 HMBOX1 ENST00000397358.3 Homeobox containing 1 MYLIP ENST00000349606.4 Myosin regulatory light chain interacting protein SEC62 ENST00000337002.4 SEC62 homolog (S. cerevisiae) TMCC1 ENST00000432054.2 Transmembrane and coiled-coil domain family 1 PHACTR4 ENST00000373839.3 Phosphatase and actin regulator 4 AIDA ENST00000340020.6 Axin interactor, dorsalization associated BEAN1 ENST00000299694.8 Brain expressed, associated with NEDD4, 1 AMMECR1 ENST00000262844.5 Alport syndrome, midface hypoplasia and elliptocytosis chro- mosomal region gene 1 EIF3J ENST00000261868.5 Eukaryotic translation initiation factor 3, subunit J SMARCA5 ENST00000283131.3 SWI/SNF related, actin dependent regulator of chromatin, subfamily a, member 5 TCF12 ENST00000267811.5 Transcription factor 12 PNO1 ENST00000263657.2 Partner of NOB1 homolog (S. cerevisiae) ZBTB37 ENST00000367701.5 Zinc finger and BTB domain containing 37 FOS ENST00000303562.4 FBJ murine osteosarcoma viral oncogene homolog ZNF385A ENST00000551109.1 Zinc -

Supplementary Table 3: Genes Only Influenced By
Supplementary Table 3: Genes only influenced by X10 Illumina ID Gene ID Entrez Gene Name Fold change compared to vehicle 1810058M03RIK -1.104 2210008F06RIK 1.090 2310005E10RIK -1.175 2610016F04RIK 1.081 2610029K11RIK 1.130 381484 Gm5150 predicted gene 5150 -1.230 4833425P12RIK -1.127 4933412E12RIK -1.333 6030458P06RIK -1.131 6430550H21RIK 1.073 6530401D06RIK 1.229 9030607L17RIK -1.122 A330043C08RIK 1.113 A330043L12 1.054 A530092L01RIK -1.069 A630054D14 1.072 A630097D09RIK -1.102 AA409316 FAM83H family with sequence similarity 83, member H 1.142 AAAS AAAS achalasia, adrenocortical insufficiency, alacrimia 1.144 ACADL ACADL acyl-CoA dehydrogenase, long chain -1.135 ACOT1 ACOT1 acyl-CoA thioesterase 1 -1.191 ADAMTSL5 ADAMTSL5 ADAMTS-like 5 1.210 AFG3L2 AFG3L2 AFG3 ATPase family gene 3-like 2 (S. cerevisiae) 1.212 AI256775 RFESD Rieske (Fe-S) domain containing 1.134 Lipo1 (includes AI747699 others) lipase, member O2 -1.083 AKAP8L AKAP8L A kinase (PRKA) anchor protein 8-like -1.263 AKR7A5 -1.225 AMBP AMBP alpha-1-microglobulin/bikunin precursor 1.074 ANAPC2 ANAPC2 anaphase promoting complex subunit 2 -1.134 ANKRD1 ANKRD1 ankyrin repeat domain 1 (cardiac muscle) 1.314 APOA1 APOA1 apolipoprotein A-I -1.086 ARHGAP26 ARHGAP26 Rho GTPase activating protein 26 -1.083 ARL5A ARL5A ADP-ribosylation factor-like 5A -1.212 ARMC3 ARMC3 armadillo repeat containing 3 -1.077 ARPC5 ARPC5 actin related protein 2/3 complex, subunit 5, 16kDa -1.190 activating transcription factor 4 (tax-responsive enhancer element ATF4 ATF4 B67) 1.481 AU014645 NCBP1 nuclear cap -

Gene-Based and Pathway-Based Genome-Wide Association Study of Alcohol Dependence
• 111 • Shanghai Archives of Psychiatry, 2015, Vol. 27, No. 2 •Original research article• Gene-based and pathway-based genome-wide association study of alcohol dependence Lingjun ZUO1, Clarence K. ZHANG2,3*, Frederick G. SAYWARD4,5, Kei-Hoi CHEUNG4, Kesheng WANG6, John H. KRYSTAL1, Hongyu ZHAO2, Xingguang LUO1* Background: The organization of risk genes within signaling pathways may provide clues about the converging neurobiological effects of risk genes for alcohol dependence. Aims: Identify risk genes and risk gene pathways for alcohol dependence. Methods: We conducted a pathway-based genome-wide association study (GWAS) of alcohol dependence using a gene-set-rich analytic approach. Approximately one million genetic markers were tested in the discovery sample which included 1409 European-American (EA) alcohol dependent individuals and 1518 EA healthy comparison subjects. An additional 681 African-American (AA) cases and 508 AA healthy subjects served as the replication sample. Results: We identified several genome-wide replicable risk genes and risk pathways that were significantly associated with alcohol dependence. After applying the Bonferroni correction for multiple testing, the ‘cell- extracellular matrix interactions’ pathway (p<2.0E-4 in EAs) and the PXN gene (which encodes paxillin) (p=3.9E-7 in EAs) within this pathway were the most promising risk factors for alcohol dependence. There were also two nominally replicable pathways enriched in alcohol dependence-related genes in both EAs (0.015≤p≤0.035) and AAs (0.025≤p≤0.050): the ‘Na+/Cl- dependent neurotransmitter transporters’ pathway and the ‘other glycan degradation’ pathway. Conclusions: These findings provide new evidence highlighting several genes and biological signaling processes that may be related to the risk for alcohol dependence. -

Cross-Species Comparison of Orthologous Gene Expression In
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2011 Cross-species comparison of orthologous gene expression in human bladder cancer and carcinogen-induced rodent models Yan Lu Washington University School of Medicine in St. Louis Pengyuan Liu Washington University School of Medicine in St. Louis Weidong Wen Washington University School of Medicine in St. Louis Clinton J. Grubbs University of Alabama - Birmingham Reid R. Townsend Washington University School of Medicine in St. Louis See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Lu, Yan; Liu, Pengyuan; Wen, Weidong; Grubbs, Clinton J.; Townsend, Reid R.; Malone, James P.; Lubet, Ronald A.; and You, Ming, ,"Cross-species comparison of orthologous gene expression in human bladder cancer and carcinogen-induced rodent models." American Journal of Translational Research.3,1. 8-27. (2011). https://digitalcommons.wustl.edu/open_access_pubs/3077 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Yan Lu, Pengyuan Liu, Weidong Wen, Clinton J. Grubbs, Reid R. Townsend, James P. Malone, Ronald A. Lubet, and Ming You This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/3077 Am J Transl Res 2011;3(1):8-27 www.ajtr.org /AJTR1008001 Original Article Cross-species comparison of orthologous gene expression in human bladder cancer and carcinogen-induced rodent models Yan Lu1,5, Pengyuan Liu1,5, Weidong Wen1, Clinton J Grubbs2, Reid R Townsend3, James P. -

1 SUPPLEMENTAL DATA Figure S1. Poly I:C Induces IFN-Β Expression
SUPPLEMENTAL DATA Figure S1. Poly I:C induces IFN-β expression and signaling. Fibroblasts were incubated in media with or without Poly I:C for 24 h. RNA was isolated and processed for microarray analysis. Genes showing >2-fold up- or down-regulation compared to control fibroblasts were analyzed using Ingenuity Pathway Analysis Software (Red color, up-regulation; Green color, down-regulation). The transcripts with known gene identifiers (HUGO gene symbols) were entered into the Ingenuity Pathways Knowledge Base IPA 4.0. Each gene identifier mapped in the Ingenuity Pathways Knowledge Base was termed as a focus gene, which was overlaid into a global molecular network established from the information in the Ingenuity Pathways Knowledge Base. Each network contained a maximum of 35 focus genes. 1 Figure S2. The overlap of genes regulated by Poly I:C and by IFN. Bioinformatics analysis was conducted to generate a list of 2003 genes showing >2 fold up or down- regulation in fibroblasts treated with Poly I:C for 24 h. The overlap of this gene set with the 117 skin gene IFN Core Signature comprised of datasets of skin cells stimulated by IFN (Wong et al, 2012) was generated using Microsoft Excel. 2 Symbol Description polyIC 24h IFN 24h CXCL10 chemokine (C-X-C motif) ligand 10 129 7.14 CCL5 chemokine (C-C motif) ligand 5 118 1.12 CCL5 chemokine (C-C motif) ligand 5 115 1.01 OASL 2'-5'-oligoadenylate synthetase-like 83.3 9.52 CCL8 chemokine (C-C motif) ligand 8 78.5 3.25 IDO1 indoleamine 2,3-dioxygenase 1 76.3 3.5 IFI27 interferon, alpha-inducible