Supplementary Materials
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Plant & Animal Genome V
January 9-13, 2016 PLANT & ANIMAL GENOME XXIV Town & Country Hotel THE INTERNATIONAL CONFERENCE San Diego, CA ON THE STATUS OF PLANT & ANIMAL GENOME RESEARCH FINAL PROGRAM & EXHIBIT GUIDE Organizing Committee Chairman: Stephen R. Heller, NIST (USA) Ī PLANT COORGANIZERS Juan F. Medrano, University of California, Dave Clements, Johns Hopkins University, Davis, USA USA Huaijun Zhou, University of California, Catherine Feuillet, Bayer CropScience, USA Davis, USA J. Perry Gustafson, University of Missouri, (Retired ), USA Ī ABSTRACT & WEBSITE Jerome P. Miksche, Emeritus Director, COORDINATORS USDA, Plant Genome Program, USA David Grant, USDA/ARS/CICGR, USA Graham Moore, John Innes Centre, UK Gerard Lazo, USDA/ARS/WRRC, USA Susan R. Wessler, University of California, Victoria Carollo Blake, USA Riverside, USA Rod A. Wing, University of Arizona, USA; Ī TRAVEL GRANTS COORDINATOR International Rice Research Institute, Tom Blake, Professor Emeritus, Philippines Montana State University, USA Ī ANIMAL COORGANIZERS Ī SPECIAL DUTY COORDINATORS Daniel Ciobanu, University of Nebraska – Hans Cheng, USDA/ARS, USA Lincoln, USA Max Rothschild, Iowa State University, USA Kwan-Suk Kim, Chungbuk National University, South Korea Sponsors and Supporters ORGANIZER Scherago International Ī USDA, Agricultural Research Service 111 Town Square Place Ī USDA, National Agricultural Library Suite 1208 USDA, National Institute of Food and Agriculture Ī Jersey City, NJ 07310 Ī John Innes Centre Phone: (201) 653-4777 Fax: (201) 653-5705 Cover artwork provided by Applied Biosystems. Originally developed for the company’s “Genetic Harvest” Agriculture Seminars, this image Email: [email protected] represents the importance of molecular genetic approaches in plant and animal research. Website: www.intlpag.org About Frasergen Frasergen is an innovative leader in cutting-edge bioinformatics and overseas in high-throughput genome sequencing, big genome data genomics. -

Protein Analysis by Mass Spectrometry
Protein Analysis by Mass Spectrometry Benjamin Taylor Barnhill Salem, Virginia B.S., Chemistry, Hampden-Sydney College, 2008 A Dissertation presented to the Graduate Faculty of the University of Virginia in Candidacy for the Degree of Doctor of Philosophy Department of Chemistry University of Virginia August, 2017 Preface I Abstract This dissertation describes two projects using mass spectrometry to analyze proteins. In project one we use LC-MS/MS and electron transfer dissociation to investigate the relationship between two Arabidopsis thaliana proteins, RGA and SPY, which are key plant growth regulators. SPY had previously been identified as an O-GlcNAc transferase that modified RGA, but our work led to the discovery that SPY is in fact a novel O-fucose transferase. Furthermore, we have established that O-GlcNAc and O-fucose modification have opposing effects on RGA. Modification with O-GlcNAc represses the growth inhibiting function of RGA, while O-fucose enhances it. The second project describes the use of mass spectrometry to identify proteins that selectively bind to regions of DNA, called somatic hypermutation (SHM) enhancer regions, shown to be necessary for targeting SHM to the Immunoglobulin gene variable region in B cells. SHM is a key step in the immune system’s generation of high affinity antibodies, but how the mutational process is confined to the immunoglobulin locus is not yet understood. Using a label free quantitation strategy we identify a number of nuclear proteins from the DT40 and Ramos B cell lines, in particular the transcription factors Ikaros and Aiolos, that preferentially bind to enhancer DNA vs. control DNA. -

Exosomal Signaling During Hypoxia Mediates Microvascular Endothelial Cell Migration and Vasculogenesis
Exosomal Signaling during Hypoxia Mediates Microvascular Endothelial Cell Migration and Vasculogenesis Carlos Salomon1*, Jennifer Ryan1, Luis Sobrevia1,2, Miharu Kobayashi1, Keith Ashman1, Murray Mitchell1, Gregory E. Rice1 1 University of Queensland Centre for Clinical Research, Herston, Queensland, Australia, 2 Cellular and Molecular Physiology Laboratory (CMPL), Division of Obstetrics and Gynaecology, School of Medicine, Faculty of Medicine, Pontificia Universidad Cato´lica de Chile, Santiago, Chile Abstract Vasculogenesis and angiogenesis are critical processes in fetal circulation and placental vasculature development. Placental mesenchymal stem cells (pMSC) are known to release paracrine factors (some of which are contained within exosomes) that promote angiogenesis and cell migration. The aims of this study were: to determine the effects of oxygen tension on the release of exosomes from pMSC; and to establish the effects of pMSC-derived exosomes on the migration and angiogenic tube formation of placental microvascular endothelial cells (hPMEC). pMSC were isolated from placental villi (8–12 weeks of gestation, n = 6) and cultured under an atmosphere of 1%, 3% or 8% O2. Cell-conditioned media were collected and exosomes (exo-pMSC) isolated by differential and buoyant density centrifugation. The dose effect (5–20 mg exosomal protein/ml) of pMSC-derived exosomes on hPMEC migration and tube formation were established using a real-time, live-cell imaging system (IncucyteTM). The exosome pellet was resuspended in PBS and protein content was established by mass spectrometry (MS). Protein function and canonical pathways were identified using the PANTHER program and Ingenuity Pathway Analysis, respectively. Exo-pMSC were identified, by electron microscopy, as spherical vesicles, with a typical cup- shape and diameters around of 100 nm and positive for exosome markers: CD63, CD9 and CD81. -

C-Jun N-Terminal Kinase Primes Endothelial Cells at Atheroprone Sites for Apoptosis
c-Jun N-terminal kinase primes endothelial cells at atheroprone sites for apoptosis Hera Ashraf Chaudhury Cardiovascular Science Unit, Imperial College London PhD ~ 1 ~ ACKNOWLEDGEMENTS Firstly, I would like to thank my supervisor Dr Paul Evans for his invaluable time, support and guidance throughout the course of my PhD. I would also like to thank him for his help and understanding during the writing and submission of this thesis. I would also like to thank everyone in the Cardiovascular Medicine Unit for providing an excellent and enjoyable environment for a novice scientist to achieve this level of credibility. I would particularly like to express my gratitude to Dr Graeme M Birdsey and Dr Joseph J. Boyle for their technical advice and especially to Mr Mustafa Zakkar for all his contribution; it has been a real pleasure to work with you. An extra special thanks to Danuta, Jay, Karine, Gunhild, Le, Shahir, Simon and Kim who made the bad days easier to deal with - thanks for all your support. I also wish to acknowledge Adam Platt (UCB Celltech, Cambridge, UK) for kindly providing the small molecule inhibitors of JNK and p38, Dr Jayne Dennis (Microarray Centre, Hammersmith Hospital, UK) for her invaluable assistance with the microarray set-up, Professor Roger Davis, (University of Massachusetts) for providing JNK1 knockout mice and Dr Andrew Clark, (Imperial College London) for supplying MKP-1 knockout mice. Finally, I would like to thank my parents, my siblings; Haseeb and Nayab and the entire family for all their love and support during the course of the PhD, especially my mother for all the late night drives to the lab to set-up extremely important experiments and Faisal for his understanding during the thesis writing stages. -

Rubinstein-Taybi Syndrome: a 10-Year Follow-Up
22nd SSBP International Research Symposium Back to Basics in Behavioural Phenotypes: Insights from Developing a Detailed Understanding of Behaviour Programme Book 4th – 6th September 2019 • Birmingham, UK Save the date! 23rd SSBP International Research Symposium will be held in Oslo, Norway in September 2020 Abstract submission opens: 1st April 2020 Registration opens: 1st May 2020 Deadline for online abstract submission: 22nd May 2020 Deadline for discounted early bird registration: 31st July 2020 Educational Day: 10th September 2020 Research Symposium: 11th – 12th September 2020 Join us in Oslo, Norway for our 23rd Research symposium, the theme will be Developmental disorders and behavioural phenotypes across the lifespan See www.ssbp.org.uk for further information nd 22 Internationaland SSBP details Research on Symposium how to submit an abstract for an oral or poster presentation The Society for the Study of Behavioural Phenotypes 4th– 6th September 2019 The 22nd SSBP International Research Symposium Back to Basics in Behavioural Phenotypes: Insights from Developing a Detailed Understanding of Behaviour Birmingham, UK 4th – 6th September 2019, Birmingham, UK 22nd International SSBP Research Symposium Contents Contents Welcome from the Conference Organisers .......................................................................................................................10 Birmingham Conference Organisers ......................................................................................................................................11 -
Supporting Figure Legend Figure S1
Electronic Supplementary Material (ESI) for Metallomics This journal is © The Royal Society of Chemistry 2013 1 Supporting Figure Legend 2 3 Figure S1: RPS4Y1 mRNA levels. Levels of this mRNA were measured as part of the validation of Affymetrix gene expression data from DEG.C. Cclones are controls (untreated 4 Beas-2B that grew in soft agar) and Vclones are clones transformed by chronic NaVO3 exposure. 5 6 Figure S1 7 8 9 Table S1: Differentially expressed genes in transformed clones (DEG.C). Gene Symbol Gene Description Adjusted Fold p-value change TMEM47 transmembrane protein 47 1.9E-02 36.196 SPTLC3 serine palmitoyltransferase, long chain base subunit 3 7.8E-03 33.565 RPS4Y1 ribosomal protein S4, Y-linked 1 6.9E-03 28.724 EYA4 eyes absent homolog 4 (Drosophila) 3.9E-03 18.298 EIF1AY eukaryotic translation initiation factor 1A, Y-linked 8.6E-03 13.872 EDIL3 EGF-like repeats and discoidin I-like domains 3 1.3E-02 12.147 DDX3Y DEAD (Asp-Glu-Ala-Asp) box polypeptide 3, Y-linked 3.9E-03 12.110 UTY ubiquitously transcribed tetratricopeptide repeat gene, Y-linked 9.5E-03 10.134 CYP1B1 cytochrome P450, family 1, subfamily B, polypeptide 1 2.2E-02 9.194 CD24 CD24 molecule 1.4E-02 8.328 1 Electronic Supplementary Material (ESI) for Metallomics This journal is © The Royal Society of Chemistry 2013 GABRA3 gamma-aminobutyric acid (GABA) A receptor, alpha 3 1.7E-02 7.643 ZNF730 zinc finger protein 730 2.0E-02 7.283 RARB|LOC100130354 retinoic acid receptor, beta | hypothetical protein LOC100130354 3.3E-02 7.168 DNER delta/notch-like EGF repeat containing -

A Network Biology Approach to Breast and Colorectal Cancers
A network biology approach to breast and colorectal cancers Rodrigo Arroyo Sánchez ADVERTIMENT. La consulta d’aquesta tesi queda condicionada a l’acceptació de les següents condicions d'ús: La difusió d’aquesta tesi per mitjà del servei TDX (www.tdx.cat) i a través del Dipòsit Digital de la UB (diposit.ub.edu) ha estat autoritzada pels titulars dels drets de propietat intel·lectual únicament per a usos privats emmarcats en activitats d’investigació i docència. No s’autoritza la seva reproducció amb finalitats de lucre ni la seva difusió i posada a disposició des d’un lloc aliè al servei TDX ni al Dipòsit Digital de la UB. No s’autoritza la presentació del seu contingut en una finestra o marc aliè a TDX o al Dipòsit Digital de la UB (framing). Aquesta reserva de drets afecta tant al resum de presentació de la tesi com als seus continguts. En la utilització o cita de parts de la tesi és obligat indicar el nom de la persona autora. ADVERTENCIA. La consulta de esta tesis queda condicionada a la aceptación de las siguientes condiciones de uso: La difusión de esta tesis por medio del servicio TDR (www.tdx.cat) y a través del Repositorio Digital de la UB (diposit.ub.edu) ha sido autorizada por los titulares de los derechos de propiedad intelectual únicamente para usos privados enmarcados en actividades de investigación y docencia. No se autoriza su reproducción con finalidades de lucro ni su difusión y puesta a disposición desde un sitio ajeno al servicio TDR o al Repositorio Digital de la UB. -

123165.F1.Pdf
Supplementary TABLE 1. The core set of 522 DEGs in the frontal cortex of AD overlapping between two cohorts identified by RNA-Seq data analysis of SRA060752 Fold Change Fold Change q_Value q_Value Entrez Gene ID Gene Symbol Gene Name Chromosome Locus (AD vs NC: (AD vs NC: (EMU) (UKY) EMU) UKY) 6863 TAC1 tachykinin, precursor 1 chr7:97361270-97369784 0.028013577 0.00232257 0.189297859 0.000638422 5121 PCP4 Purkinje cell protein 4 chr21:41239346-41301322 0.060477403 0.00232257 0.263070248 0.000638422 793 CALB1 calbindin 1, 28kDa chr8:91070837-91095107 0.068756027 0.00232257 0.229478357 0.000638422 54112 GPR88 G protein-coupled receptor 88 chr1:101002397-101008223 0.083735314 0.00232257 0.380695653 0.000638422 143162 FRMPD2 FERM and PDZ domain containing 2 chr10:49364156-49482941 0.086014876 0.00232257 0.344904773 0.00248927 6588 SLN sarcolipin chr11:107578100-107582787 0.091647542 0.00398798 0.110806929 0.00396642 6750 SST somatostatin chr3:187386693-187388201 0.094938693 0.00232257 0.042406817 0.000638422 63974 NEUROD6 neurogenic differentiation 6 chr7:31377079-31380538 0.095418314 0.00232257 0.159354316 0.000638422 3358 HTR2C 5-hydroxytryptamine (serotonin) receptor 2C chrX:113818550-114144627 0.09625865 0.00232257 0.211803744 0.000638422 771 CA12 carbonic anhydrase XII chr15:63615729-63674075 0.104187926 0.00232257 0.453231488 0.0218249 7432 VIP vasoactive intestinal peptide chr6:153053815-153080902 0.117494082 0.00232257 0.404586476 0.0139553 400120 SERTM1 serine-rich and transmembrane domain containing 1 chr13:37248048-37272564 0.130991037 -
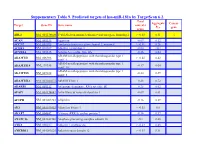
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Dissertation Alexander Kanitz
Research Collection Doctoral Thesis Tools and strategies for the unraveling of post-transcriptional gene regulatory networks Author(s): Kanitz, Alexander Publication Date: 2012 Permanent Link: https://doi.org/10.3929/ethz-a-7358945 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library DISS. ETH Nr. 20147 TOOLS AND STRATEGIES FOR THE UNRAVELING OF POST-TRANSCRIPTIONAL GENE REGULATORY NETWORKS A dissertation submitted to ETH ZURICH for the degree of Doctor of Sciences presented by ALEXANDER KANITZ M.Sc. University of Amsterdam born November 15, 1980 citizen of Germany Accepted on the recommendation of Prof. Dr. Michael Detmar Prof. Dr. Jonathan Hall Prof. Dr. André Gerber 2012 1 SUMMARY _________________________________________________________________________________ 6 1.1 Summary _____________________________________________________________________________ 6 1.2 Zusammenfassung ______________________________________________________________________ 9 2 INTRODUCTION ________________________________________________________________________ 12 2.1 Key principles and players of post-transcriptional gene regulatory processes ____________________ 13 2.1.1 The fate of eukaryotic messenger RNAs _______________________________________________ 13 2.1.2 Cis -regulatory elements ____________________________________________________________ 15 2.1.3 Trans -acting factors _______________________________________________________________ -

WO 2011/050470 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date _ . ... , . _ . 5 May 2011 (05.05.2011) WO 2011/050470 Al (51) International Patent Classification: Master University, 1200 Main Street West, MDCL 5029, C12N 5/071 (2010.01) C12Q 1/02 (2006.01) Hamilton, Ontario L8N 3Z5 (CA). RAMPALLI-DESH- A61K 35/12 (2006.01) C12Q 1/68 (2006.01) PANDE, Shravanti [IN/CA]; Stem Cell and Cancer Re C12N 15/00 (2006.01) C12N 15/867 (2006.01) search Institute, McMaster University, 1200 Main Street West, MDCL5029, Hamilton, Ontario L8N 3Z5 (CA). (21) International Application Number: MUNOZ RISUENO, Ruth [ES/CA]; Stem Cell and PCT/CA20 10/00 1708 Cancer Research Institute, McMaster University, 1200 (22) International Filing Date: Main Street West, MDCL5029, Hamilton, Ontario L8N 29 October 2010 (29.10.2010) 3Z5 (CA). VIJAYARAGAVAN, Kausalia [CA/ES]; Stem Cell and Cancer Research Institute, McMaster Uni (25) Filing Language: English versity, 1200 Main Street West, MDCL5029, Hamilton, (26) Publication Language: English Ontario L8N 3Z5 (CA). (30) Priority Data: (74) Agent: BERESKIN & PARR LLP/S.E.N.C.R.L., 61/256,1 70 29 October 2009 (29.10.2009) US S.R.L.; 40 King Street West, 40th Floor, Toronto, On tario M5H 3Y2 (CA). (71) Applicant (for all designated States except US): MC¬ MASTER UNIVERSITY [CA/CA]; McMaster Industry (81) Designated States (unless otherwise indicated, for every Liaison Office (MILO), McMaster Innovation Park, 175 kind of national protection available): AE, AG, AL, AM, Longwood Road South, Room 305, Hamilton, Ontario AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, L8P 0A1 (CA).