Development of EST-Molecular Markers from RNA Sequencing for Genetic Management and Identification of Growth Traits in Potato Grouper (Epinephelus Tukula)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Checklist of Serranid and Epinephelid Fishes (Perciformes: Serranidae & Epinephelidae) of India
Journal of the Ocean Science Foundation 2021, Volume 38 Checklist of serranid and epinephelid fishes (Perciformes: Serranidae & Epinephelidae) of India AKHILESH, K.V. 1, RAJAN, P.T. 2, VINEESH, N. 3, IDREESBABU, K.K. 4, BINEESH, K.K. 5, MUKTHA, M. 6, ANULEKSHMI, C. 1, MANJEBRAYAKATH, H. 7, GLADSTON, Y. 8 & NASHAD M. 9 1 ICAR-Central Marine Fisheries Research Institute, Mumbai Regional Station, Maharashtra, India. Corresponding author: [email protected]; Email: [email protected] 2 Andaman & Nicobar Regional Centre, Zoological Survey of India, Port Blair, India. Email: [email protected] 3 Department of Health & Family Welfare, Government of West Bengal, India. Email: [email protected] 4 Department of Science and Technology, U.T. of Lakshadweep, Kavaratti, India. Email: [email protected] 5 Southern Regional Centre, Zoological Survey of India, Chennai, Tamil Nadu, India. Email: [email protected] 6 ICAR-Central Marine Fisheries Research Institute, Visakhapatnam Regional Centre, Andhra Pradesh, India. Email: [email protected] 7 Centre for Marine Living Resources and Ecology, Kochi, Kerala, India. Email: [email protected] 8 ICAR-Central Island Agricultural Research Institute, Port Blair, Andaman and Nicobar Islands, India. Email: [email protected] 9 Fishery Survey of India, Port Blair, Andaman and Nicobar Islands, 744101, India. Email: [email protected] Abstract We provide an updated checklist of fishes of the families Serranidae and Epinephelidae reported or listed from India, along with photographs. A total of 120 fishes in this group are listed as occurring in India based on published literature, of which 25 require further confirmation and validation. We confirm here the presence of at least 95 species in 22 genera occurring in Indian marine waters. -

Fact Sheet: Potato Cod
Fact Sheet: Potato Cod Region North Coast, Gascoyne Coast Summary The captivating potato cod is truly a giant of the fish kingdom. Its massive size and homebody nature draws divers who are looking for a story to tell about their underwater adventure. Figure 1. Potato cod (Image: © Shannon Conway) Scientific name Epinephelus tukula Distribution Generated on 25/09/2021 https://marinewaters.fish.wa.gov.au/resource/potato-cod/ Page 1 of 4 Widely distributed in Indo-Pacific tropical and subtropical waters from the Red Sea to the Solomon Islands. In Western Australia from Exmouth to Wyndham. Ecological suite PROTECTED SPECIES General description The captivating potato cod is truly a giant of the fish kingdom. Its massive size and homebody nature draws divers who are looking for a story to tell about their underwater adventure. Potato cod have been known to get super friendly with divers who visit regularly, even growing accustomed to hand feeding. It is these features that make the potato cod highly susceptible to overfishing. As a result, the species is irregular in its distribution and not often seen in areas open to fishing. Rather, it is in protected areas such as marine parks, reserves and sanctuary zones that the species thrives. An ambush predator, the potato cod hides behind coral bombies waiting for prey to pass. When spotted, it lunges from its hiding spot, consuming in a single gulp, its prey. Generated on 25/09/2021 https://marinewaters.fish.wa.gov.au/resource/potato-cod/ Page 2 of 4 Figure 2. Potato cod identifying features (Image: Department of Fisheries) The Rowley Shoals are a well known haunt for potato cod – be sure to check the channels and coral outcrops if lucky enough to be in the water at this spectacular marine destination! Other names Potato rockcod, grouper, potato bass, potato grouper Habitat Coral reef habitats exposed to currents, such as channels or seamounts. -

NORTH COAST FISH IDENTIFICATION GUIDE Ben M
NORTH COAST FISH IDENTIFICATION GUIDE Ben M. Rome and Stephen J. Newman Department of Fisheries 3rd floor SGIO Atrium 168-170 St George’s Terrace PERTH WA 6000 Telephone (08) 9482 7333 Facsimile (08) 9482 7389 Website: www.fish.wa.gov.au ABN: 55 689 794 771 Published by Department of Fisheries, Perth, Western Australia. Fisheries Occasional Publications No. 80, September 2010. ISSN: 1447 - 2058 ISBN: 1 921258 90 X Information about this guide he intention of the North Coast Fish Identification Guide is to provide a simple, Teasy to use manual to assist commercial, recreational, charter and customary fishers to identify the most commonly caught marine finfish species in the North Coast Bioregion. This guide is not intended to be a comprehensive taxonomic fish ID guide for all species. It is anticipated that this guide will assist fishers in providing a more comprehensive species level description of their catch and hence assist scientists and managers in understanding any variation in the species composition of catches over both spatial and temporal scales. Fish taxonomy is a dynamic and evolving field. Advances in molecular analytical techniques are resolving many of the relationships and inter-relationships among species, genera and families of fishes. In this guide, we have used and adopted the latest taxonomic nomenclature. Any changes to fish taxonomy will be updated and revised in subsequent editions. The North Coast Bioregion extends from the Ashburton River near Onslow to the Northern Territory border. Within this region there is a diverse range of habitats from mangrove creeks, rivers, offshore islands, coral reef systems to continental shelf and slope waters. -
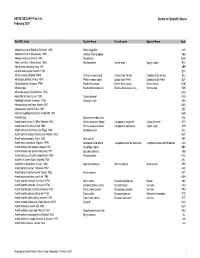
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Sexual Maturation and Gonad Development in Tiger Grouper (Epinephelus Fuscoguttatus) X Giant Grouper (E
e Rese tur arc ul h c & a u D q e A v e f l o o Luin et al., J Aquac Res Development 2013, 5:2 l p a m n Journal of Aquaculture r e u n o t DOI: 10.4172/2155-9546.1000213 J ISSN: 2155-9546 Research & Development Research Article OpenOpen Access Access Sexual Maturation and Gonad Development in Tiger Grouper (Epinephelus Fuscoguttatus) X Giant Grouper (E. Lanceolatus) Hybrid Marianne Luin*, Ching Fui Fui and Shigeharu Senoo Borneo Marine Research Institute, Universiti Malaysia Sabah,Malaysia. Abstract The objective of this study is to determine the possibility of sexual maturation of tiger grouper x giant grouper (TGGG) hybrid. Specimens of TGGG were reared in the hatchery for six years in 150-tonne tanks equipped with a water recirculation system. Observations on maturation were conducted. TGGG (49 specimens) were measured for their total length, standard length, head length, body height, body width, body circumference and body weight, which were 73.97 ± 5.69 cm; 62.09 ± 5.10 cm; 22.87 ± 2.06 cm; 22.84 ± 2.42 cm; 13.98 ± 1.74 cm; 58.94 ± 6.18 cm; 9.88 ± 2.46 kg, respectively. Cannulation method could not be done for 80% of the population for TGGG hybrid grouper. The condition factor of TGGG averaged 2.40 ± 0.21 (n=49). Length-weight relationship of TGGG showed a strong correlation (P>0.05) and the equation obtained were: log W = -4.3317 + 2.8453 log L. The value of regression coefficient (b) equals to 2.8453 and value of correlation coefficient (r) equals to 0.93. -
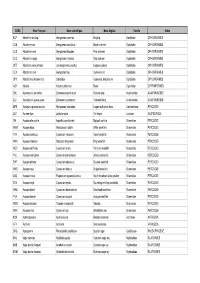
Nom Français
CODE Nom Français Nom scientifique Nom Anglais Famille Ordre KCP Abadèche du Cap Genypterus capensis Kingklip Ophidiidae OPHIDIIFORMES CUB Abadèche noir Genypterus maculatus Black cusk-eel Ophidiidae OPHIDIIFORMES CUS Abadèche rosé Genypterus blacodes Pink cusk-eel Ophidiidae OPHIDIIFORMES CUC Abadèche rouge Genypterus chilensis Red cusk-eel Ophidiidae OPHIDIIFORMES OFZ Abadèche sans jambes Lamprogrammus exutus Legless cuskeel Ophidiidae OPHIDIIFORMES CEX Abadèches nca Genypterus spp Cusk-eels nei Ophidiidae OPHIDIIFORMES OPH Abadèches, brotules nca Ophidiidae Cusk-eels, brotulas nei Ophidiidae OPHIDIIFORMES ALR Ablette Alburnus alburnus Bleak Cyprinidae CYPRINIFORMES ZML Acanthure à pierreries Zebrasoma gemmatum Spotted tang Acanthuridae ACANTHUROIDEI ZLV Acanthure à queue jaune Zebrasoma xanthurum Yellowtail tang Acanthuridae ACANTHUROIDEI MPS Achigan à grande bouche Micropterus salmoides Largemouth black bass Centrarchidae PERCOIDEI LQT Acmée râpe Lottia limatula File limpet Lottiidae GASTROPODA ISA Acoupa aile-courte Isopisthus parvipinnis Bigtooth corvina Sciaenidae PERCOIDEI WEW Acoupa blanc Atractoscion nobilis White weakfish Sciaenidae PERCOIDEI YNV Acoupa cambucu Cynoscion virescens Green weakfish Sciaenidae PERCOIDEI WKK Acoupa chasseur Macrodon ancylodon King weakfish Sciaenidae PERCOIDEI WEP Acoupa du Pérou Cynoscion analis Peruvian weakfish Sciaenidae PERCOIDEI YNJ Acoupa mongolare Cynoscion jamaicensis Jamaica weakfish Sciaenidae PERCOIDEI SWF Acoupa pintade Cynoscion nebulosus Spotted weakfish Sciaenidae PERCOIDEI WKS Acoupa -
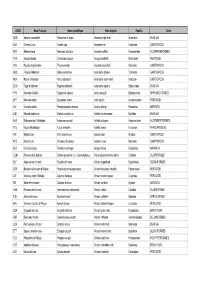
Liste Espèces
CODE Nom Français Nom scientifique Nom Anglais Famille Ordre ODQ Anomie cascabelle Pododesmus cepio Abalone jingle shell Anomiidae BIVALVIA ABX Ormeaux nca Haliotis spp Abalones nei Haliotidae GASTROPODA REN Sébaste rose Sebastes fasciatus Acadian redfish Scorpaenidae SCORPAENIFORMES YNA Acoupa toeroe Cynoscion acoupa Acoupa weakfish Sciaenidae PERCOIDEI HSV Pourpre aiguillonnee Thais aculeata Aculeate rock shell Muricidae GASTROPODA GBQ Troque d'Adanson Gibbula adansoni Adanson's gibbula Trochidae GASTROPODA NKA Natice d'Adanson Natica adansoni Adanson's moon snail Naticidae GASTROPODA GLW Tagal d'Adanson Tagelus adansonii Adanson's tagelus Solecurtidae BIVALVIA PYD Manchot d'Adélie Pygoscelis adeliae Adelie penguin Spheniscidae SPHENISCIFORMES QFT Maconde aden Synagrops adeni Aden splitfin Acropomatidae PERCOIDEI NIV Crevette adonis Parapenaeopsis venusta Adonis shrimp Penaeidae NATANTIA DJD Modiole adriatique Modiolus adriaticus Adriatic horse mussel Mytilidae BIVALVIA AAA Esturgeon de l'Adriatique Acipenser naccarii Adriatic sturgeon Acipenseridae ACIPENSERIFORMES FCV Fucus d'Adriatique Fucus virsoides Adriatic wrack Fucaceae PHAEOPHYCEAE IRR Mitre brûlée Mitra eremitarum Adusta miter Mitridae GASTROPODA KCE Murex bruni Chicoreus brunneus Adusta murex Muricidae GASTROPODA AES Crevette ésope Pandalus montagui Aesop shrimp Pandalidae NATANTIA CGM Poisson-chat, hybride Clarias gariepinus x C. macrocephalus Africa-bighead catfish, hybrid Clariidae SILURIFORMES SUF Ange de mer africain Squatina africana African angelshark Squatinidae SQUALIFORMES -

Checklist of the Shore Fishes of Europa Island, Mozambique Channel, Southwestern Indian Ocean, Including 302 New Records
Stuttgarter Beiträge zur Naturkunde A, Neue Serie 6: 247–276; Stuttgart, 30.IV.2013 247 Checklist of the shore fishes of Europa Island, Mozambique Channel, southwestern Indian Ocean, including 302 new records RONALD FRICKE, PATRICK DURVILLE, GIACOMO BERNARDI, PHILIPPE BORSA, GÉRARD MOU-THAM & PASCALE CHABANET Abstract An annotated checklist of the fish species of Europa Island (Mozambique Channel, southwestern Indian Ocean) comprises a total of 389 species in 62 families. 302 species are recorded from Europa Island for the first time. All species are autochthonous; no introduced species have been found. The fish fauna is exclusively marine, with the Labridae, Pomacentridae, Serranidae, Gobiidae and Acanthuridae being the families with most representatives. The fish fauna at Europa Island is typical for offshore, low islands in the southwestern Indian Ocean. Zoogeo- graphically, the main element of the fish fauna of Europa Island consists of widespread tropical Indo-Pacific species (292 species, 75.1 % of the total occurring species). A total of 13 species (3.3 %) are found worldwide, either cir- cumtropical or circumtropical including warm temperate zones. An additional 76 species (19.5 %) are Indian Ocean endemics, including 36 western Indian Ocean endemics (9.2 %), and 10 southwestern Indian Ocean endemics (2.6 %). No endemic fish species are reported for Europa Island. K e y w o r d s : Checklist, Pisces, southwestern Indian Ocean, Europa Island, new records, zoogeography. Zusammenfassung Die Checkliste der Fische der Insel Europa (Kanal von Mosambik, südwestlicher Indischer Ozean) enthält 389 Arten in 62 Familien. 302 Arten werden zum ersten Mal von der Insel Europa beobachtet. Alle Fischarten sind au- thochthon; es wurden keine durch den Menschen eingeführten oder eingeschleppten Arten gefunden. -

Studies on the Reproductive Biology of the Female Spiny Cheek Grouper, Epinephelus Diacanthus (Valenciennes, 1828)
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by CMFRI Digital Repository Indian J. Fish., 56(2) : 87-94, 2009 87 Studies on the reproductive biology of the female spiny cheek grouper, Epinephelus diacanthus (Valenciennes, 1828) A. CHANDRASEKHARA RAO AND L. KRISHNAN Central Marine Fisheries Research Institute, P. O. Box. 1603, Cochin - 682 018, Kerala, India e-mail: [email protected] ABSTRACT Morphological and histological examination of gonads and detailed classification and description of maturity stages were carried out in the female Epinephelus diacanthus. Maximum ova diameter of 650 µm was observed in the ripe ovary. Oocyte size increased with the maturation of gonads. Gonado-somatic Index (GSI) values also increased with the maturation of gonads. Fecundity was in the range of 13.1 x 103 to 145.7 x 103. Keywords: Epinephelus diacanthus, Gonado-somatic index, Reproductive biology, Spiny cheek grouper Introduction 502 mm (Tessy, 1994; James et al., 1996). Eventhough many studies were carried out on brood stock development Reproduction is a dynamic metabolic activity in most of Epinephelus tauvina and Epinephelus malabaricus fishes and it involves sequential changes in the germ cells. (Mathew et al., 2002; Mathew, 2005), little information is The pattern of these changes in the gonads is typical for available on E. diacanthus which forms a dominant share each species. To clearly understand the physiology of fish in grouper capture fisheries (Chakraborthy, 1994). during reproduction, the study of the seasonal developmental changes of gonads through both In order to proceed with the artificial means of macroscopic and microscopic observations is necessary. -

6. Index of Scientific and Vernacular Names
click for previous page 356 FAO Species Catalogue Vol. 16 6. INDEX OF SCIENTIFIC AND VERNACULAR NAMES EXPLANATION OF THE SYSTEM Type faces used : Italics (bold) : Valid scientific names (double entry by genera and species) Italics : Synonyms (double entry by genera and species) Roman (bold) : International (FAO) species names Roman : Local species names Groupers of the World 357 amblycephalus, Epinephelus ...........................109-110 A smblycephalus, Serranus ....................................... 109 Abade .................................................................. 265 amboinensis, Serranus .......................................... 250 Abadèch .............................................................. 135 lAnak keretang .................................................... 110 Abadejo .............................................................. 265 lAnak pertang .....................................................110 Abadejo ............................................................... 267 lanalis, Cephalopholis ................................... 36, 58-59 Abae ................................................................... 265 lanalis, Serranus ..................................................... 35 abdominalis, Serranus ............................................ 174 lanalogus, Epinephelus ............. 53, 69,103,110,148,174 acanthistius, Bodianus .................................. 69-70, 102 landersoni, Epinephelus ..................................111-112 acanthistius, Cephalopholis .................................... -

Oma158267.Pdf
ب Fishery Quality Control Regulation 12/2009 Third Edition 2009 ت 5 ث ج Contents Chapter Page Chapter (One) Definitions 8 Chapter (Two) General provisions 12 Chapter (Three) General conditions for quality control and safety of fish and 20 fishery products Chapter (Four) Additional conditions for chilled fish and fishery production 24 Chapter (Five) Additional conditions for frozen fish and fishery products 26 Chapter (Six) Additional conditions for chilled and frozen crustaceans 27 Chapter (Seven) Potable water inspection and test methods 29 Chapter (Eight) General conditions for displaying and marketing of fish and 29 fishery products Chapter (Nine) Hygienic conditions for the production and marketing of fish and 31 fishery products Chapter (Ten) Requirements during and after fish and fishery products landing 33 Chapter (Eleven) Conditions for establishment on land 36 Chapter (Twelve) Conditions for handling fish and fishery production on shore 39 Chapter (Thirteen) Hygienic conditions for monitoring fish and fishery products 45 production. Chapter (Fourteen) Violations and Penalties 48 Annexes Annex No. (1) .............................................................................................. 50 Annex No. (2) .............................................................................................. 57 Annex No. (3) .............................................................................................. 67 Annex No. (4) .............................................................................................. 72 -
Reef Fishes Addressing Challenges to Coastal Ecosystem and Livelihood Issues
Field Guide to About Mangroves for the Future Field Guide to Mangroves for the Future (MFF) is a unique partner-led initiative to promote investment in coastal ecosystem conservation for sustainable development. Co-chaired by IUCN and UNDP, MFF provides a platform for collaboration among the many different agencies, sectors and countries which are Reef Fishes addressing challenges to coastal ecosystem and livelihood issues. The goal is to promote an integrated ocean-wide approach to coastal management Reef Fishes and to building the resilience of ecosystem-dependent coastal communities. MFF builds on a history of coastal management interventions before and after the 2004 Indian Ocean tsunami. It initially focused on the countries that were of Sri Lanka worst affected by the tsunami — India, Indonesia, Maldives, Seychelles, Sri Lanka and Thailand. More recently it has expanded to include Bangladesh, of Sri Lanka Cambodia, Pakistan and Viet Nam. Mangroves are the flagship of the initiative, but MFF is inclusive of all types of coastal ecosystems, such as coral reefs, estuaries, lagoons, sandy beaches, sea grasses and wetlands. The MFF grants facility offers small, medium and large grants to support initiatives that provide practical, hands-on demonstrations of effective coastal management in action. Each country manages its own MFF programme through a National Coordinating Body which includes representation from government, NGOs and the private sector. Vol.2 MFF addresses priorities for long-term sustainable coastal ecosystem man- agement which include, among others: climate change adaptation and miti- gation, disaster risk reduction, promotion of ecosystem health, development of sustainable livelihoods, and active engagement of the private sector in developing sustainable business practices.