Environmental DNA Reveals the Fine-Grained and Hierarchical
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Marine Ecology Progress Series 477:177
Vol. 477: 177–188, 2013 MARINE ECOLOGY PROGRESS SERIES Published March 12 doi: 10.3354/meps10144 Mar Ecol Prog Ser Larval exposure to shared oceanography does not cause spatially correlated recruitment in kelp forest fishes Jenna M. Krug*, Mark A. Steele Department of Biology, California State University, 18111 Nordhoff St., Northridge, California 91330-8303, USA ABSTRACT: In organisms that have a life history phase whose dispersal is influenced by abiotic forcing, if individuals of different species are simultaneously exposed to the same forcing, spa- tially correlated settlement patterns may result. Such correlated recruitment patterns may affect population and community dynamics. The extent to which settlement or recruitment is spatially correlated among species, however, is not well known. We evaluated this phenomenon among 8 common kelp forest fishes at 8 large reefs spread over 30 km of the coast of Santa Catalina Island, California. In addition to testing for correlated recruitment, we also evaluated the influences of predation and habitat quality on spatial patterns of recruitment. Fish and habitat attributes were surveyed along transects 7 times during 2008. Using these repeated surveys, we also estimated the mortality rate of the prey species that settled most consistently (Oxyjulis californica) and evaluated if mortality was related to recruit density, predator density, or habitat attributes. Spatial patterns of recruitment of the 8 study species were seldom correlated. Recruitment of all species was related to one or more attributes of the habitat, with giant kelp abundance being the most widespread predictor of recruitment. Mortality of O. californica recruits was density-dependent and declined with increasing canopy cover of giant kelp, but was unrelated to predator density. -

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

California Saltwater Sport Fishing Regulations
2017–2018 CALIFORNIA SALTWATER SPORT FISHING REGULATIONS For Ocean Sport Fishing in California Effective March 1, 2017 through February 28, 2018 13 2017–2018 CALIFORNIA SALTWATER SPORT FISHING REGULATIONS Groundfish Regulation Tables Contents What’s New for 2017? ............................................................. 4 24 License Information ................................................................ 5 Sport Fishing License Fees ..................................................... 8 Keeping Up With In-Season Groundfish Regulation Changes .... 11 Map of Groundfish Management Areas ...................................12 Summaries of Recreational Groundfish Regulations ..................13 General Provisions and Definitions ......................................... 20 General Ocean Fishing Regulations ��������������������������������������� 24 Fin Fish — General ................................................................ 24 General Ocean Fishing Fin Fish — Minimum Size Limits, Bag and Possession Limits, and Seasons ......................................................... 24 Fin Fish—Gear Restrictions ................................................... 33 Invertebrates ........................................................................ 34 34 Mollusks ............................................................................34 Crustaceans .......................................................................36 Non-commercial Use of Marine Plants .................................... 38 Marine Protected Areas and Other -

CHECKLIST and BIOGEOGRAPHY of FISHES from GUADALUPE ISLAND, WESTERN MEXICO Héctor Reyes-Bonilla, Arturo Ayala-Bocos, Luis E
ReyeS-BONIllA eT Al: CheCklIST AND BIOgeOgRAphy Of fISheS fROm gUADAlUpe ISlAND CalCOfI Rep., Vol. 51, 2010 CHECKLIST AND BIOGEOGRAPHY OF FISHES FROM GUADALUPE ISLAND, WESTERN MEXICO Héctor REyES-BONILLA, Arturo AyALA-BOCOS, LUIS E. Calderon-AGUILERA SAúL GONzáLEz-Romero, ISRAEL SáNCHEz-ALCántara Centro de Investigación Científica y de Educación Superior de Ensenada AND MARIANA Walther MENDOzA Carretera Tijuana - Ensenada # 3918, zona Playitas, C.P. 22860 Universidad Autónoma de Baja California Sur Ensenada, B.C., México Departamento de Biología Marina Tel: +52 646 1750500, ext. 25257; Fax: +52 646 Apartado postal 19-B, CP 23080 [email protected] La Paz, B.C.S., México. Tel: (612) 123-8800, ext. 4160; Fax: (612) 123-8819 NADIA C. Olivares-BAñUELOS [email protected] Reserva de la Biosfera Isla Guadalupe Comisión Nacional de áreas Naturales Protegidas yULIANA R. BEDOLLA-GUzMáN AND Avenida del Puerto 375, local 30 Arturo RAMíREz-VALDEz Fraccionamiento Playas de Ensenada, C.P. 22880 Universidad Autónoma de Baja California Ensenada, B.C., México Facultad de Ciencias Marinas, Instituto de Investigaciones Oceanológicas Universidad Autónoma de Baja California, Carr. Tijuana-Ensenada km. 107, Apartado postal 453, C.P. 22890 Ensenada, B.C., México ABSTRACT recognized the biological and ecological significance of Guadalupe Island, off Baja California, México, is Guadalupe Island, and declared it a Biosphere Reserve an important fishing area which also harbors high (SEMARNAT 2005). marine biodiversity. Based on field data, literature Guadalupe Island is isolated, far away from the main- reviews, and scientific collection records, we pres- land and has limited logistic facilities to conduct scien- ent a comprehensive checklist of the local fish fauna, tific studies. -

Fish Bulletin No. 109. the Barred Surfperch (Amphistichus Argenteus Agassiz) in Southern California
UC San Diego Fish Bulletin Title Fish Bulletin No. 109. The Barred Surfperch (Amphistichus argenteus Agassiz) in Southern California Permalink https://escholarship.org/uc/item/9fh0623k Authors Carlisle, John G, Jr. Schott, Jack W Abramson, Norman J Publication Date 1960 eScholarship.org Powered by the California Digital Library University of California STATE OF CALIFORNIA DEPARTMENT OF FISH AND GAME MARINE RESOURCES OPERATIONS FISH BULLETIN No. 109 The Barred Surfperch (Amphistichus argenteus Agassiz) in Southern Califor- nia By JOHN G. CARLISLE, JR., JACK W. SCHOTT and NORMAN J. ABRAMSON 1960 1 2 3 4 ACKNOWLEDGMENTS The Surf Fishing Investigation received a great deal of help in the conduct of its field work. The arduous task of beach seining all year around was shared by many members of the California State Fisheries Laboratory staff; we are particularly grateful to Mr. Parke H. Young and Mr. John L. Baxter for their willing and continued help throughout the years. Mr. Frederick B. Hagerman was project leader for the first year of the investigation, until his recall into the Air Force, and he gave the project an excellent start. Many others gave help and advice, notably Mr. John E. Fitch, Mr. Phil M. Roedel, Mr. David C. Joseph, and Dr. F. N. Clark of this laboratory. Dr. Carl L. Hubbs of Scripps Institution of Oceanography at La Jolla gave valuable advice, and we are indebted to the late Mr. Conrad Limbaugh of the same institution for accounts of his observations on surf fishes, and for SCUBA diving instructions. The project was fortunate in securing able seasonal help, particularly from Mr. -
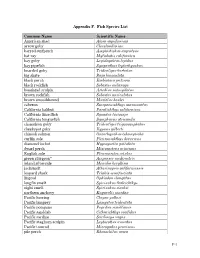
Appendix E: Fish Species List
Appendix F. Fish Species List Common Name Scientific Name American shad Alosa sapidissima arrow goby Clevelandia ios barred surfperch Amphistichus argenteus bat ray Myliobatis californica bay goby Lepidogobius lepidus bay pipefish Syngnathus leptorhynchus bearded goby Tridentiger barbatus big skate Raja binoculata black perch Embiotoca jacksoni black rockfish Sebastes melanops bonehead sculpin Artedius notospilotus brown rockfish Sebastes auriculatus brown smoothhound Mustelus henlei cabezon Scorpaenichthys marmoratus California halibut Paralichthys californicus California lizardfish Synodus lucioceps California tonguefish Symphurus atricauda chameleon goby Tridentiger trigonocephalus cheekspot goby Ilypnus gilberti chinook salmon Oncorhynchus tshawytscha curlfin sole Pleuronichthys decurrens diamond turbot Hypsopsetta guttulata dwarf perch Micrometrus minimus English sole Pleuronectes vetulus green sturgeon* Acipenser medirostris inland silverside Menidia beryllina jacksmelt Atherinopsis californiensis leopard shark Triakis semifasciata lingcod Ophiodon elongatus longfin smelt Spirinchus thaleichthys night smelt Spirinchus starksi northern anchovy Engraulis mordax Pacific herring Clupea pallasi Pacific lamprey Lampetra tridentata Pacific pompano Peprilus simillimus Pacific sanddab Citharichthys sordidus Pacific sardine Sardinops sagax Pacific staghorn sculpin Leptocottus armatus Pacific tomcod Microgadus proximus pile perch Rhacochilus vacca F-1 plainfin midshipman Porichthys notatus rainwater killifish Lucania parva river lamprey Lampetra -

(Teleostei: Gobiidae) Ensemble Along of Eastern Tropical Pacific: Biological Inventory, Latitudinal Variation and Species Turnover
RESEARCH ARTICLE Gamma-diversity partitioning of gobiid fishes (Teleostei: Gobiidae) ensemble along of Eastern Tropical Pacific: Biological inventory, latitudinal variation and species turnover Omar Valencia-MeÂndez1☯, FabiaÂn Alejandro RodrõÂguez-Zaragoza2☯, Luis Eduardo Calderon-Aguilera3☯¤, Omar DomõÂnguez-DomõÂnguez4☯, AndreÂs LoÂpez-PeÂrez5☯* a1111111111 1 Doctorado en Ciencias BioloÂgicas y de la Salud, Universidad AutoÂnoma Metropolitana-Iztapalapa, Ciudad de MeÂxico, MeÂxico, 2 Departamento de EcologõÂa, CUCBA, Universidad de Guadalajara, Jalisco, MeÂxico, a1111111111 3 Departamento de EcologõÂa Marina, Centro de InvestigacioÂn CientõÂfica y de EducacioÂn Superior de a1111111111 Ensenada (CICESE), Ensenada, Baja California, MeÂxico, 4 Facultad de BiologõÂa, Universidad Michoacana a1111111111 de San NicolaÂs de Hidalgo, Morelia, MichoacaÂn, MeÂxico, 5 Departamento de HidrobiologõÂa, Universidad a1111111111 AutoÂnoma Metropolitana-Iztapalapa, Ciudad de MeÂxico, MeÂxico ☯ These authors contributed equally to this work. ¤ Current address: University of Southampton, Southampton, United Kingdom * [email protected] OPEN ACCESS Citation: Valencia-MeÂndez O, RodrõÂguez-Zaragoza Abstract FA, Calderon-Aguilera LE, DomõÂnguez-DomÂõnguez O, LoÂpez-PeÂrez A (2018) Gamma-diversity Gobies are the most diverse marine fish family. Here, we analysed the gamma-diversity partitioning of gobiid fishes (Teleostei: Gobiidae) ensemble along of Eastern Tropical Pacific: (γ-diversity) partitioning of gobiid fishes to evaluate the additive and multiplicative compo- Biological inventory, latitudinal variation and nents of α and β-diversity, species replacement and species loss and gain, at four spatial species turnover. PLoS ONE 13(8): e0202863. scales: sample units, ecoregions, provinces and realms. The richness of gobies from the https://doi.org/10.1371/journal.pone.0202863 realm Eastern Tropical Pacific (ETP) is represented by 87 species. Along latitudinal and lon- Editor: Heather M. -

Terrestrial and Marine Biological Resource Information
APPENDIX C Terrestrial and Marine Biological Resource Information Appendix C1 Resource Agency Coordination Appendix C2 Marine Biological Resources Report APPENDIX C1 RESOURCE AGENCY COORDINATION 1 The ICF terrestrial biological team coordinated with relevant resource agencies to discuss 2 sensitive biological resources expected within the terrestrial biological study area (BSA). 3 A summary of agency communications and site visits is provided below. 4 California Department of Fish and Wildlife: On July 30, 2020, ICF held a conference 5 call with Greg O’Connell (Environmental Scientist) and Corianna Flannery (Environmental 6 Scientist) to discuss Project design and potential biological concerns regarding the 7 Eureka Subsea Fiber Optic Cables Project (Project). Mr. O’Connell discussed the 8 importance of considering the western bumble bee. Ms. Flannery discussed the 9 importance of the hard ocean floor substrate and asked how the cable would be secured 10 to the ocean floor to reduce or eliminate scour. The western bumble bee has been 11 evaluated in the Biological Resources section of the main document, and direct and 12 indirect impacts are avoided. The Project Description describes in detail how the cable 13 would be installed on the ocean floor, the importance of the hard bottom substrate, and 14 the need for avoidance. 15 Consultation Outcomes: 16 • The Project was designed to avoid hard bottom substrate, and RTI Infrastructure 17 (RTI) conducted surveys of the ocean floor to ensure that proper routing of the 18 cable would occur. 19 • Ms. Flannery will be copied on all communications with the National Marine 20 Fisheries Service 21 California Department of Fish and Wildlife: On August 7, 2020, ICF held a conference 22 call with Greg O’Connell to discuss a site assessment and survey approach for the 23 western bumble bee. -

Redescription and Taxonomic Status of Dipturus Chilensis (Guichenot, 1848), and Description of Dipturus Lamillai Sp
Zootaxa 4590 (5): 501–524 ISSN 1175-5326 (print edition) https://www.mapress.com/j/zt/ Article ZOOTAXA Copyright © 2019 Magnolia Press ISSN 1175-5334 (online edition) https://doi.org/10.11646/zootaxa.4590.5.1 http://zoobank.org/urn:lsid:zoobank.org:pub:F484C560-CAE9-4A9E-B408-AEC2C8893DAD Redescription and taxonomic status of Dipturus chilensis (Guichenot, 1848), and description of Dipturus lamillai sp. nov. (Rajiformes: Rajidae), a new species of long-snout skate from the Falkland Islands FRANCISCO J. CONCHA1,2,7, JANINE N. CAIRA1, DAVID A. EBERT3,4,5 & JOOST H. W. POMPERT6 1Department of Ecology & Evolutionary Biology, University of Connecticut, 75 North Eagleville Road, Unit 3043 Storrs, CT 06269 – 3043, USA 2Facultad de Ciencias del Mar y de Recursos Naturales, Universidad de Valparaíso, Av. Borgoño 16344, Viña del Mar, Chile 3Pacific Shark Research Center, Moss Landing Marine Laboratories, Moss Landing, CA 95039, USA 4Department of Ichthyology, California Academy of Sciences, 55 Music Concourse Drive, San Francisco, CA 94118, USA 5South African Institute for Aquatic Biodiversity, Private Bag 1015, Grahamstown 6140, South Africa 6Georgia Seafoods Ltd, Waverley House, Stanley, Falkland Islands, United Kingdom 7Corresponding author. E-mail: [email protected] Abstract Recent molecular evidence has called into question the identity of skates collected in the waters off the Falkland Islands previously identified as Zearaja chilensis. NADH2 sequence data indicate that these specimens are not conspecific with those currently referred to as Z. chilensis from Chile and, in fact, represent a novel cryptic species. This study aimed to investigate this hypothesis based on morphological comparisons of specimens from the coasts of both western and eastern South America. -

2019 Annual Marine Environmental Analysis and Interpretation Report
2019 ANNUAL MARINE ENVIRONMENTAL ANALYSIS AND INTERPRETATION San Onofre Nuclear Generating Station ANNUAL MARINE ENVIRONMENTAL ANALYSIS AND INTERPRETATION San Onofre Nuclear Generating Station July 2020 Page intentionally blank Report Preparation/Data Collection – Oceanography and Marine Biology MBC Aquatic Sciences 3000 Red Hill Avenue Costa Mesa, CA 92626 Page intentionally blank TABLE OF CONTENTS LIST OF FIGURES ........................................................................................................................... iii LIST OF TABLES ...............................................................................................................................v LIST OF APPENDICES ................................................................................................................... vi EXECUTIVE SUMMARY .............................................................................................................. vii CHAPTER 1 STUDY INTRODUCTION AND GENERATING STATION DESCRIPTION 1-1 INTRODUCTION ................................................................................................................... 1-1 PURPOSE OF SAMPLING ......................................................................................... 1-1 REPORT APPROACH AND ORGANIZATION ........................................................ 1-1 DESCRIPTION OF THE STUDY AREA ................................................................... 1-1 HISTORICAL BACKGROUND ........................................................................................... -

A List of Common and Scientific Names of Fishes from the United States And
t a AMERICAN FISHERIES SOCIETY QL 614 .A43 V.2 .A 4-3 AMERICAN FISHERIES SOCIETY Special Publication No. 2 A List of Common and Scientific Names of Fishes -^ ru from the United States m CD and Canada (SECOND EDITION) A/^Ssrf>* '-^\ —---^ Report of the Committee on Names of Fishes, Presented at the Ei^ty-ninth Annual Meeting, Clearwater, Florida, September 16-18, 1959 Reeve M. Bailey, Chairman Ernest A. Lachner, C. C. Lindsey, C. Richard Robins Phil M. Roedel, W. B. Scott, Loren P. Woods Ann Arbor, Michigan • 1960 Copies of this publication may be purchased for $1.00 each (paper cover) or $2.00 (cloth cover). Orders, accompanied by remittance payable to the American Fisheries Society, should be addressed to E. A. Seaman, Secretary-Treasurer, American Fisheries Society, Box 483, McLean, Virginia. Copyright 1960 American Fisheries Society Printed by Waverly Press, Inc. Baltimore, Maryland lutroduction This second list of the names of fishes of The shore fishes from Greenland, eastern the United States and Canada is not sim- Canada and the United States, and the ply a reprinting with corrections, but con- northern Gulf of Mexico to the mouth of stitutes a major revision and enlargement. the Rio Grande are included, but those The earlier list, published in 1948 as Special from Iceland, Bermuda, the Bahamas, Cuba Publication No. 1 of the American Fisheries and the other West Indian islands, and Society, has been widely used and has Mexico are excluded unless they occur also contributed substantially toward its goal of in the region covered. In the Pacific, the achieving uniformity and avoiding confusion area treated includes that part of the conti- in nomenclature. -

Humboldt Bay Fishes
Humboldt Bay Fishes ><((((º>`·._ .·´¯`·. _ .·´¯`·. ><((((º> ·´¯`·._.·´¯`·.. ><((((º>`·._ .·´¯`·. _ .·´¯`·. ><((((º> Acknowledgements The Humboldt Bay Harbor District would like to offer our sincere thanks and appreciation to the authors and photographers who have allowed us to use their work in this report. Photography and Illustrations We would like to thank the photographers and illustrators who have so graciously donated the use of their images for this publication. Andrey Dolgor Dan Gotshall Polar Research Institute of Marine Sea Challengers, Inc. Fisheries And Oceanography [email protected] [email protected] Michael Lanboeuf Milton Love [email protected] Marine Science Institute [email protected] Stephen Metherell Jacques Moreau [email protected] [email protected] Bernd Ueberschaer Clinton Bauder [email protected] [email protected] Fish descriptions contained in this report are from: Froese, R. and Pauly, D. Editors. 2003 FishBase. Worldwide Web electronic publication. http://www.fishbase.org/ 13 August 2003 Photographer Fish Photographer Bauder, Clinton wolf-eel Gotshall, Daniel W scalyhead sculpin Bauder, Clinton blackeye goby Gotshall, Daniel W speckled sanddab Bauder, Clinton spotted cusk-eel Gotshall, Daniel W. bocaccio Bauder, Clinton tube-snout Gotshall, Daniel W. brown rockfish Gotshall, Daniel W. yellowtail rockfish Flescher, Don american shad Gotshall, Daniel W. dover sole Flescher, Don stripped bass Gotshall, Daniel W. pacific sanddab Gotshall, Daniel W. kelp greenling Garcia-Franco, Mauricio louvar