Protein Domain-Based Prediction of Compound–Target Interactions And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Atnea1-Identification and Characterization of a Novel Plant Nuclear Envelope Associated Protein
Vol. 13(7), pp. 844-856, 12 February, 2014 DOI: 10.5897/AJB2013.13078 ISSN 1684-5315 ©2014 Academic Journals African Journal of Biotechnology http://www.academicjournals.org/AJB Full Length Research Paper AtNEA1-identification and characterization of a novel plant nuclear envelope associated protein Ting Lu Department of Biological and Medical Sciences, Oxford Brookes University, Oxford OX3 0BP, UK. Accepted 29 January, 2014 In animal and yeast cells, a cross nuclear envelope structure linker of nucleoskeleton and cytoskeleton (LINC) is formed by outer nuclear membrane SUN proteins and inner nuclear membrane KASH proteins. However, little information was acquired about plant SUN-KASH structure until they were found in plant SUN proteins in 2010 and KASH proteins in 2012. The SUN-KASH complex alongside with actin in the microfilament cytoskeleton and nucleaoskeleton together shape the main cell skeleton structure and involve in many important biological functions including cell structure stability, cell movement and cell division. In searching of other plant nuclear envelope associated proteins, arabidopsis nuclear envelop associated (AtNEA1) protein 1, a plant nucleoplasmic protein, was identified from biological studies. AtNEA1 was predicted to have a nuclear localisation signal (NLS), two coiled coil domains and one transmembrane (TM) domain. The mutants with the deletion of respective putative domains were observed under confocal microscopy. The subcellular localisation of mutants implied that putative NLS is not essential for AtNEA1 to diffuse through NPC but can strongly increase the efficiency, both coiled coil domains participate in the interaction of AtNEA1 with its unknown INM intrinsic interaction partner, and putative TM appeared to be non-functional. -

Identification of Plant SUN Domain-Interacting Tail Proteins and Analysis of Their Function in Nuclear Positioning
Identification of Plant SUN Domain-Interacting Tail Proteins and Analysis of Their Function in Nuclear Positioning DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of the Ohio State University By Xiao Zhou, M. S. Graduate Program in Plant Cellular and Molecular Biology The Ohio State University 2013 Dissertation Committee: Professor Iris Meier, Advisor Professor Biao Ding Professor Stephen Osmani Professor R. Keith Slotkin Copyright by Xiao Zhou 2013 Abstract The nuclear envelope (NE) is a double membrane system consisting of an inner nuclear membrane (INM) and an outer nuclear membrane (ONM). Studies in opisthokonts revealed that the two membranes are bridged by protein complexes formed by the INM Sad1/UNC-84 (SUN) proteins and the ONM Klarsicht/ANC-1/Syne-1 homology (KASH) proteins. These SUN-KASH NE bridges are usually linkers of the nucleoskeleton to the cytoskeleton (LINC) conserved across eukaryotes. LINC complexes are key players in multiple cellular processes, such as nuclear and chromosomal positioning and nuclear shape determination, which in turn influence the gametogenesis and several aspects of development. Although these cellular processes have long been also known in plants, no KASH proteins are encoded in the plant genomes. I identified WPP domain interacting proteins (WIPs) as the first plant KASH protein analogs. WIPs are plant-specific ONM proteins that redundantly anchor Ran GTPase activating protein (RanGAP) to the NE. Arabidopsis thaliana WIPs (AtWIPs) interact with Arabidopsis thaliana SUN proteins (AtSUNs), which is required for both AtWIP1 and AtRanGAP1 NE localization. In addition, AtWIPs and AtSUNs are necessary for maintaining the elongated nuclear shape of Arabidopsis epidermal cells. -

Nesprins: from the Nuclear Envelope and Beyond
expert reviews http://www.expertreviews.org/ in molecular medicine Nesprins: from the nuclear envelope and beyond Dipen Rajgor and Catherine M. Shanahan* Nuclear envelope spectrin-repeat proteins (Nesprins), are a novel family of nuclear and cytoskeletal proteins with rapidly expanding roles as intracellular scaffolds and linkers. Originally described as proteins that localise to the nuclear envelope (NE) and establish nuclear-cytoskeletal connections, nesprins have now been found to comprise a diverse spectrum of tissue specific isoforms that localise to multiple sub-cellular compartments. Here, we describe how nesprins are necessary in maintaining cellular architecture by acting as essential scaffolds and linkers at both the NE and other sub-cellular domains. More importantly, we speculate how nesprin mutations may disrupt tissue specific nesprin scaffolds and explain the tissue specific nature of many nesprin-associated diseases, including laminopathies. The eukaryotic cytoplasm contains three major composed of three α-helical bundles with a left- types of cytoskeletal filaments: Filamentous- handed twist, and its primary function is to actin (F-actin), microtubules (MTs) and provide docking sites for proteins and other intermediate filaments (IFs). These components higher order complexes (Refs 3, 4). Although are organised in a manner that provides the cell most SR proteins contain CHDs, some possess with an internal framework fundamental for motifs which can interact with other cytoskeletal many processes, such as controlling cellular components, allowing linkage of SR-associated shape, polarity, adhesion and migration, complexes to filamentous structures other than cytokinesis, inter- and intracellular F-actin. In addition, these motifs allow cross- Nesprins: from the nuclear envelope and beyond communication and trafficking of organelles, linking between different filaments and dynamic vesicles, proteins and RNA (Refs 1, 2). -

The Nuclear Envelope: Target and Mediator of the Apoptotic Process Liora Lindenboim1, Hila Zohar1,Howardj.Worman2 and Reuven Stein1
Lindenboim et al. Cell Death Discovery (2020) 6:29 https://doi.org/10.1038/s41420-020-0256-5 Cell Death Discovery REVIEW ARTICLE Open Access The nuclear envelope: target and mediator of the apoptotic process Liora Lindenboim1, Hila Zohar1,HowardJ.Worman2 and Reuven Stein1 Abstract Apoptosis is characterized by the destruction of essential cell organelles, including the cell nucleus. The nuclear envelope (NE) separates the nuclear interior from the cytosol. During apoptosis, the apoptotic machinery, in particular caspases, increases NE permeability by cleaving its proteins, such as those of the nuclear pore complex (NPC) and the nuclear lamina. This in turns leads to passive diffusion of cytosolic apoptogenic proteins, such as caspases and nucleases, through NPCs into the nucleus and the subsequent breakdown of the NE and destruction of the nucleus. However, NE leakiness at early stages of the apoptotic process can also occur in a caspase-independent manner, where Bax, by a non-canonical action, promotes transient and repetitive localized generation and subsequent rupture of nuclear protein-filled nuclear bubbles. This NE rupture leads to discharge of apoptogenic nuclear proteins from the nucleus to the cytosol, a process that can contribute to the death process. Therefore, the NE may play a role as mediator of cell death at early stages of apoptosis. The NE can also serve as a platform for assembly of complexes that regulate the death process. Thus, the NE should be viewed as both a mediator of the cell death process and a target. 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Facts redistribution of the nuclear proteins to the cytosol that might subsequently act as amplifiers of the ● The NE is an important target of the apoptotic apoptotic process. -

Table S4. Trophoblast Differentiation-Associated Genes
Table S4. Trophoblast differentiation-associated genes Gene Stem Chromosomal Affymetrix ID Gene Title Symbol Ave Dif Ave GenBank Location dif/ stem t-test 1390511_at LOC308394 Cgm4 10 1863 BI285801 1 193.51 0.006 1378534_at similar to brain carcinoembryonic antigen LOC308394 10 1746 NM_001025679 1q21 183.10 0.001 1388433_at keratin complex 1, acidic, gene 19 Krt1-19 53 7958 NM_199498 10q32.1 149.07 0.022 1369029_at phospholipid scramblase 1 Plscr1 20 2050 NM_057194 8q31 101.12 0.028 1389856_at carcinoembryonic antigen gene family 4 Cgm4 57 5073 NM_012525 1q21 89.73 0.001 carcinoembryonic antigen-related cell 1368996_at adhesion molecule 3 Ceacam3 202 17879 NM_012702 1q21 88.71 0.001 1392832_at similar to angiopoietin-like 1 LOC684489 17 1398 XM_001068284 --- 83.66 0.001 1377666_at choline dehydrogenase Chdh 26 1571 NM_198731 16p16 60.59 0.003 cytochrome P450, family 11, subfamily a, 1368468_at polypeptide 1 Cyp11a1 196 9216 NM_017286 8q24 47.11 0.000 1376934_x_at similar to brain carcinoembryonic antigen Cgm4 53 2146 BI285801 1 40.22 0.000 stimulated by retinoic acid gene 6 homolog 1390525_a_at (mouse) Stra6 28 1037 NM_001029924 8q24 37.44 0.001 1382690_at carcinoembryonic antigen gene family 4 Cgm4 81 2729 NM_012525 1q21 33.86 0.001 1367809_at prolactin family 4, subfamily a, member 1 Prl4a1 683 22573 NM_017036 17p11 33.05 0.004 calcium channel, voltage-dependent, L type, 1383458_at alpha 1D subunit Cacna1d 26 802 BF403759 16 30.89 0.001 1370852_at spleen protein 1 precursor LOC171573 692 20968 NM_138537 8q21 30.29 0.003 1376036_at transporter -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

The Emerin-Binding Transcription Factor Lmo7 Is Regulated by Association with P130cas at Focal Adhesions
The emerin-binding transcription factor Lmo7 is regulated by association with p130Cas at focal adhesions Michele A. Wozniak1,2 , Brendon M. Baker2, Christopher S. Chen2 and Katherine L. Wilson1 1 Department of Cell Biology, Johns Hopkins University School of Medicine, Baltimore, MD, USA 2 Department of Bioengineering, University of Pennsylvania, Philadelphia, PA, USA ABSTRACT Loss of function mutations in the nuclear inner membrane protein, emerin, cause X-linked Emery-Dreifuss muscular dystrophy (X-EDMD). X-EDMD is character- ized by contractures of major tendons, skeletal muscle weakening and wasting, and cardiac conduction system defects. The transcription factor Lmo7 regulates muscle- and heart-relevant genes and is inhibited by binding to emerin, suggesting Lmo7 misregulation contributes to EDMD disease. Lmo7 associates with cell adhesions and shuttles between the plasma membrane and nucleus, but the regulation and biological consequences of this dual localization were unknown. We report endoge- nous Lmo7 also associates with focal adhesions in cells, and both co-localizes and co-immunoprecipitates with p130Cas, a key signaling component of focal adhesions. Lmo7 nuclear localization and transcriptional activity increased significantly in p130Cas-null MEFs, suggesting Lmo7 is negatively regulated by p130Cas-dependent association with focal adhesions. These results support EDMD models in which Lmo7 is a downstream mediator of integrin-dependent signaling that allows tendon cells and muscles to adapt to and withstand mechanical stress. Subjects Submitted 14 April 2013 Biochemistry, Cell Biology, Molecular Biology Accepted 29 July 2013 Keywords Lmo7, p130Cas, Focal adhesions, Emery-Dreifuss muscular dystrophy, Laminopathy, Published 20 August 2013 LEM-domain, Nuclear envelope, Nucleoskeleton, Tendon, Emerin Corresponding author Katherine L. -

Sharmin Supple Legend 150706
Supplemental data Supplementary Figure 1 Generation of NPHS1-GFP iPS cells (A) TALEN activity tested in HEK 293 cells. The targeted region was PCR-amplified and cloned. Deletions in the NPHS1 locus were detected in four clones out of 10 that were sequenced. (B) PCR screening of human iPS cell homologous recombinants (C) Southern blot screening of human iPS cell homologous recombinants Supplementary Figure 2 Human glomeruli generated from NPHS1-GFP iPS cells (A) Morphological changes of GFP-positive glomeruli during differentiation in vitro. A different aggregate from the one shown in Figure 2 is presented. Lower panels: higher magnification of the areas marked by rectangles in the upper panels. Note the shape changes of the glomerulus (arrowheads). Scale bars: 500 µm. (B) Some, but not all, of the Bowman’s capsule cells were positive for nephrin (48E11 antibody: magenta) and GFP (green). Scale bars: 10 µm. Supplementary Figure 3 Histology of human podocytes generated in vitro (A) Transmission electron microscopy of the foot processes. Lower magnification of Figure 4H. Scale bars: 500 nm. (B) (C) The slit diaphragm between the foot processes. Higher magnification of the 1 regions marked by rectangles in panel A. Scale bar: 100 nm. (D) Absence of mesangial or vascular endothelial cells in the induced glomeruli. Anti-PDGFRβ and CD31 antibodies were used to detect the two lineages, respectively, and no positive signals were observed in the glomeruli. Podocytes are positive for WT1. Nuclei are counterstained with Nuclear Fast Red. Scale bars: 20 µm. Supplementary Figure 4 Cluster analysis of gene expression in various human tissues (A) Unbiased cluster analysis across various human tissues using the top 300 genes enriched in GFP-positive podocytes. -

Connecting the Nucleus to the Cytoskeleton by SUN–KASH
COCEBI-1074; NO. OF PAGES 6 Available online at www.sciencedirect.com Connecting the nucleus to the cytoskeleton by SUN–KASH bridges across the nuclear envelope Erin C Tapley and Daniel A Starr The nuclear–cytoskeleton connection influences many aspects with a wide variety of cytoskeletal components [11]. of cellular architecture, including nuclear positioning, the Mutations in mammalian SUN and KASH proteins lead stiffness of the global cytoskeleton, and mechanotransduction. to developmental defects in neurogenesis, gametogen- Central to all of these processes is the assembly and function of esis, myogenesis, cilliogenesis, and retina formation and conserved SUN–KASH bridges, or LINC complexes, that span contribute to human diseases, including muscular dystro- the nuclear envelope. Recent studies provide details of the phy, ataxia, Progeria, lissencephaly, and cancer higher order assembly and targeting of SUN proteins to the [11,17,18,19,20 ]. inner nuclear membrane. Structural studies characterize SUN– KASH interactions that form the central link of the nuclear- The rapidly growing field of nuclear–cytoskeletal inter- envelope bridge. KASH proteins at the outer nuclear membrane actions has recently been reviewed [11–13]. Here, with link the nuclear envelope to the cytoskeleton where forces are apologies to the rest of the field, we focus on five major generated to move nuclei. Significantly, SUN proteins were findings reported over the past two years. The first step of recently shown to contribute to the progression of building the SUN–KASH bridge is recruiting SUN laminopathies. proteins to the inner nuclear membrane. Surprisingly, trafficking SUN proteins to the inner nuclear membrane Address involves multiple, partially redundant mechanisms Department of Molecular and Cellular Biology, University of California, [21 ,22 ,23 ]. -
Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection
Int. J. Mol. Sci. 2016, 17, 98; doi:10.3390/ijms17010098 S1 of S4 Supplementary Information: Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection Chun-Hou Liao, Bing-Huei Chen, Han-Sun Chiang, Chiu-Wei Chen, Mei-Feng Chen, Chih-Chun Ke, Ya-Yun Wang, Wei-Ning Lin, Chi-Chung Wang and Ying-Hung Lin Table S1. List of expressional changes of up- regulated and down-regulated genes from D-gal-injected mice. Gene Full Name Fold Up-regulated genes Gpx3 Glutathione peroxidase 3 13.1 C3 Complement component 3 8.5 H2-Aa Histocompatibility 2, class II antigen A, α 6.9 Ctss Cathepsin S 6.3 Cylc2 Cylicin, basic protein of sperm head cytoskeleton 2 5.6 H2-Eb1 Histocompatibility 2, class II antigen E β 5.2 Psmb7 Proteasome (prosome, macropain) subunit, β type 7 4.3 Frg1 FSHD region gene 1 4.2 Safb2 Scaffold attachment factor B2 4.1 Gbp2 Guanylate binding protein 2 4 B2m β-2 microglobulin 4 Tbx3 T-box 3 3.8 Snrpa Small nuclear ribonucleoprotein polypeptide A 3.7 SWI/SNF related, matrix associated, actin dependent regulator of Smarca2 3.7 Chromatin, subfamily a, member 2 Gm13237 Predicted gene 13237 3.6 Cep85l Centrosomal protein 85-like 3.5 Dcn Decorin 3.2 Nme8 NME/NM23 family member 8 3.2 Dhx9 DEAH (Asp-Glu-Ala-His) box polypeptide 9 3.2 Dnajb11 DnaJ (Hsp40) homolog, subfamily B, member 11 3.2 Sltm SAFB-like, transcription modulator 3.1 Fus Fused in sarcoma 3.1 Anxa1 Annexin A1 3.1 Ly86 Lymphocyte antigen 86 3 Hpgd Hydroxyprostaglandin dehydrogenase 15 (NAD) 3 Ccdc112 Coiled-coil domain containing 112 3 Efcab2 EF-hand calcium binding domain 2 3 C1qc Complement component 1, q subcomponent, C chain 3 Dnajc21 DnaJ (Hsp40) homolog, subfamily C, member 21 2.9 Ccl8 Chemokine (C–C motif) ligand 8 2.9 Ccdc186 Coiled-coil domain containing 186 2.9 Plagl1 Pleomorphic adenoma gene-like 1 2.8 Amy1 Amylase 1, salivary 2.8 Cdkl2 Cyclin-dependent kinase-like 2 (CDC2-related kinase) 2.8 Nucb2 Nucleobindin 2 2.8 Ifitm2 Interferon induced transmembrane protein 2 2.8 Int. -

Downloaded for Analysis
bioRxiv preprint doi: https://doi.org/10.1101/343939; this version posted June 10, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: 2 Identification and characterization of conserved and divergent genes encoding the 3 nuclear envelope LINC complex in maize (Zea mays L.). 4 5 Running Title: 6 Maize LINC complex 7 8 Authors and Affiliations: 9 Gumber, Hardeep K.1; McKenna, Joseph F.2; Estrada, Amado L.1; Tolmie, Andrea F.2 10 Graumann, Katja2; Bass, Hank W.1 11 12 1. Department of Biological Science, Florida State University, Tallahassee, FL, 13 USA, 32306-4295 14 2. Department of Biological and Medical Sciences, Faculty of Health and Life 15 Sciences, Oxford Brookes University, Oxford, UK, OX30BP. 16 17 18 Corresponding Author: 19 [email protected] 20 21 Key words: 22 LINC, SUN, KASH, nuclear envelope, maize 23 bioRxiv preprint doi: https://doi.org/10.1101/343939; this version posted June 10, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 24 SUMMARY STATEMENT 25 Genes encoding maize candidates for the core LINC and associated complex proteins 26 have been comprehensively identified with functional validation by one or more assays 27 for several of the KASH genes. 28 29 ABSTRACT 30 The LINC (Linker of Nucleoskeleton to Cytoskeleton) complex serves as an essential 31 multi-protein structure spanning the nuclear envelope. It connects the cytoplasm to the 32 nucleoplasm and functions to maintain nuclear shape and architecture, as well as 33 regulates chromosome dynamics during mitosis and meiosis. -
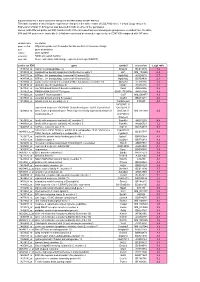
Supplementary File 1
Supplementary file 1: Gene expression changes in the white matter of CD47 KO mice This table contains a list of all gene expression changes in the white matter of CD47 KO mice ( > 2-fold: |Log2 ratio| > 1) Expression of total 14,875 genes was detected in both or either of the genotypes. Genes (with different probe set ID#) found in both of the increased and decreased gene groups were excluded from the table. 594 and 548 genes were markedly (> 2-fold) increased and decreased, respectively, in CD47 KO compared with WT mice. column name description probe set ID# Affymetrix probe set ID number for Mouse 430 2.0 Genome Arrays gene gene description symbol gene symbol accession NCBI accession number Log2 ratio Gene expression fold change expressed as Log2 (KO/WT) probe set ID# gene symbol accession Log2 ratio 1449153_at matrix metallopeptidase 12 Mmp12 BC019135 7.2 1419534_at oxidized low density lipoprotein (lectin-like) receptor 1 Olr1 NM_138648 6.2 1444176_at ATPase, H+ transporting, lysosomal V0 subunit D2 Atp6v0d2 AV204216 5.7 1434798_at ATPase, H+ transporting, lysosomal V0 subunit D2 Atp6v0d2 BB769890 2.3 1420504_at solute carrier family 6 (neurotransmitter transporter), member 14 Slc6a14 AF320226 5.5 1459934_at ubiquitin specific peptidase 42 Usp42 AA516835 5.2 1431800_at von Willebrand factor A domain containing 8 Vwa8 AK004956 5.2 1431927_at RIKEN cDNA 5033417F24 gene 5033417F24Rik AK018199 4.9 1419202_at cystatin F (leukocystatin) Cst7 NM_009977 4.9 1439943_at vacuolar protein sorting 54 (yeast) Vps54 BB201271 4.6 1419966_at tubulin,