Excited-State Dynamics in Photosystem II: Insights from the X-Ray Crystal Structure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

You Light up My Life
Chapter 7: Photosynthesis Electromagnetic Spectrum Shortest Gamma rays wavelength X-rays UV radiation Visible light Infrared radiation Microwaves Longest Radio waves wavelength Photons • Packets of light energy • Each type of photon has fixed amount of energy • Photons having most energy travel as shortest wavelength (blue-violet light) Visible Light shortest range of most radiation range of heat escaping longest wavelengths reaching Earth’s surface from Earth’s surface wavelengths (most energetic) (lowest energy) gamma x ultraviolet near-infrared infrared microwaves radio rays rays radiation radiation radiation waves VISIBLE LIGHT 400 450 500 550 600 650 700 Wavelengths of light (nanometers) • Wavelengths humans perceive as different colors • Violet (380 nm) to red (750 nm) • Longer wavelengths, lower energy Fig. 7-2, p.108 Pigments • Colors you can see are the wavelengths not absorbed • These light catching particles capture energy from the various wavelengths. Variety of Pigments Chlorophylls a and b Carotenoids - orange Anthocyanins - purple/red Phycobilins - red Xanthophylls - yellow Chlorophylls chlorophyll a chlorophyll b Wavelength absorption (%) absorption Wavelength Wavelength (nanometers) Accessory Pigments Carotenoids, Phycobilins, Anthocyanins beta-carotene phycoerythrin (a phycobilin) percent of wavelengths absorbed wavelengths (nanometers) Pigments Fig. 7-3a, p.109 Pigments Fig. 7-3b, p.109 Pigments Fig. 7-3c, p.109 Pigments Fig. 7-3d, p.109 http://www.youtube.com/watch?v=fwGcOg PB10o&feature=fvsr Fig. 7-3e, p.109 Pigments Fig. 7-3e, p.109 Pigments in Photosynthesis • Bacteria – Pigments in plasma membranes • Plants – Pigments and proteins organized in chloroplast membranes T.E. Englemann’s Experiment Background • Certain bacterial cells will move toward places where oxygen concentration is high • Photosynthesis produces oxygen T.E. -

SCHELVIS CV Profile 2010
Curriculum vitae: Johannes Schelvis 09/7/2010 PERSONAL INFORMATION Johannes P. M. Schelvis, Associate Professor Montclair State University Department of Chemistry and Biochemistry 1 Normal Avenue Montclair, NJ 07043 EDUCATION B.S., Physics, 1985, Free University, Amsterdam, Netherlands Ph.D., Biophysics, 1995, University of Leiden, Leiden, Netherlands PROFESSIONAL EXPERIENCE Associate Professor Montclair State University September 2007 – present Assistant Professor New York University September 2000 – August 2007 Postdoctoral Researcher Michigan State University March 1995 - August 2000 HONORS AND AWARDS • Institute Fellow, Margaret and Herman Sokol Institute for the Pharmaceutical Life Sciences at Montclair State University, September 2008 - present • Goddard Fellowship, New York University, 2004 • Whitehead Fellowship for Junior Faculty in Biomedical or Biological Sciences, New York University, 2003. GRANTS AWARDED ACTIVE • "Molecular Mechanisms of Photolyase and Cryptochrome" National Science Foundation, MCB-0920013, August 2009 – July 2012 , $419,453 t.c. (PI) • "Binding of ICER to Its Own Promoter as a Mode of Cooperative Regulation" Margaret and Herman Sokol Institute for Pharmaceutical Life Sciences, September 2008 – August 2011 (1-year no cost extension), $100,000 (PI with Dr. Carlos Molina) • "Light-Driven Damage and Repair of DNA", Faculty Scholarship Program, Montclair State University, 2008 – 2012 , 6 TCH (PI) COMPLETED • "Fingerprinting DNA Damage" Margaret and Herman Sokol Faculty/Student Research Grant Program, July 2008 -

Genetically Engineered Mutant of the Cyanobacterium Synechocystis
Proc. Natl. Acad. Sci. USA Vol. 83, pp. 9474-9477, December 1986 Botany Genetically engineered mutant of the cyanobacterium Synechocystis 6803 lacks the photosystem II chlorophyll-binding protein CP-47 (photosynthesis/cartridge mutagenesis/reaction center/chlorophyll fluorescence) WIM F. J. VERMAAS*t, JOHN G. K. WILLIAMS*, A. WILLIAM RUTHERFORDt, PAUL MATHISt, AND CHARLES J. ARNTZEN* *Central Research and Development Department, E. I. du Pont de Nemours and Co., Inc., Wilmington, DE 19898; and tCentre d'Etudes Nucleaires de Saclay, Departement de Biologie, Service de Biophysique, 91191 Gif-sur-Yvette Cedex, France Contributed by Charles J. Arntzen, August 29, 1986 ABSTRACT CP-47 is absent in a genetically engineered also be found in an analogous location in the D1 and D2 mutant of cyanobacterium Synechocystis 6803, in which the proteins. psbB gene [encoding the chlorophyll-binding photosystem U To obtain more information on the role of CP-47 (and (PSII) protein CP-47] was interrupted. Another chlorophyll- thereby possibly to localize P680), a well-defined mutant of binding PSI1 protein, CP-43, is present in the mutant, and the cyanobacterium Synechocystis 6803 was constructed. In functionally inactive PSII-enriched particles can be isolated this mutant (9) the only change was an interruption of the from mutant thylakoids. We interpret these data as indicating psbB gene (encoding CP-47) by a piece ofDNA containing the that the PSII core complex of the mutant still assembles in the gene for aminoglycoside-3'-phosphotransferase, conferring absence of CP-47. The mutant lacks a 77 K fluorescence kanamycin resistance. This mutant is no longer capable of emission maximum at 695 nm, suggesting that the PSII reaction PSII electron transport (9). -

Studies of the Two Redox Active Tyrosines in Photosystem II
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1527 Studies of the two redox active tyrosines in Photosystem II NIGAR AHMADOVA ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-554-9933-4 UPPSALA urn:nbn:se:uu:diva-320916 2017 Dissertation presented at Uppsala University to be publicly examined in Room 2001, Ångströmlaboratoriet, Lägerhyddsvägen 1, Uppsala, Wednesday, 14 June 2017 at 13:15 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Professor Robert Burnap (Oklahoma State University, USA). Abstract Ahmadova, N. 2017. Studies of the two redox active tyrosines in Photosystem II. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1527. 72 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-9933-4. Photosystem II is a unique enzyme which catalyzes light induced water oxidation. This process is driven by highly oxidizing ensemble of four Chl molecules, PD1, PD2, ChlD1 and ChlD2 called, + - - P680. Excitation of one of the Chls in P680 leads to the primary charge separation, P680 Pheo . Pheo transfers electrons sequentially to the primary quinone acceptor QA and the secondary quinone + acceptor QB. P680 in turn extracts electrons from Mn4CaO5 cluster, a site for the water oxidation. There are two redox active tyrosines, TyrZ and TyrD, found in PSII. They are symmetrically located on the D1 and D2 central proteins. Only TyrZ acts as intermediate electron carrier between P680 and Mn4CaO5 cluster, while TyrD does not participate in the linear electron flow and stays oxidized under light conditions. -

PHOTOSYNTHESIS Photosystem II Psba Yes Photosystem II P680
PHOTOSYNTHESIS Photosystem II PsbA yes Photosystem II P680 reaction center D1 protein PsbD yes Photosystem II P680 reaction center D2 protein PsbC yes Photosystem II CP43 chlorophyll apoprotein PsbB yes Photosystem II CP47 chlorophyll apoprotein PsbE yes Photosystem II cytochrome b559 subunit alpha PsbF yes Photosystem II cytochrome b559 subunit beta PsbL yes Photosystem II PsbL protein PsbJ yes Photosystem II PsbJ protein PsbK yes Photosystem II PsbK protein PsbM yes Photosystem II PsbM protein PsbH yes Photosystem II PsbH protein PsbI yes Photosystem II PsbI protein PsbO yes Photosystem II oxygen-evolving enhancer protein 1 PsbP yes Photosystem II oxygen-evolving enhancer protein 2 PsbQ no Photosystem II oxygen-evolving enhancer protein 3 PsbR no Photosystem II 10 kDa protein PsbS no Photosystem II 22kDa protein PsbT yes Photosystem II PsbT protein PsbU yes Photosystem II PsbU protein PsbV yes Photosystem II cytochrome c550 PsbW no Photosystem II PsbW protein PsbX yes Photosystem II PsbX protein PsbY yes Photosystem II PsbY protein PsbZ yes Photosystem II PsbZ protein Psb27 yes Photosystem II Psb27 protein Psb28 yes Photosystem II 13kDa protein Psb28-2 no Photosystem II Psb28-2 protein Photosystem I PsaA yes Photosystem I P700 chlorophyll a apoprotein A1 PsaB yes Photosystem I P700 chlorophyll a apoprotein A2 PsaC yes Photosystem I subunit VII PsaD yes Photosystem I subunit II PsaE yes Photosystem I subunit IV PsaF yes Photosystem I subunit III PsaG no Photosystem I subunit V PsaH no Photosystem I subunit VI PsaI yes Photosystem I subunit -

Photosynthesis the Electromagnetic Spectrum
BI/CH 422/622 ANABOLISM OUTLINE: Overview Key experiments: Light causes oxygen, which is from water splitting (Hill) NADPH made (Ochoa) Separate from carbohydrate biosynthesis (Rubin & Kamen) Light Reactions energy in a photon pigments HOW Light absorbing complexes Reaction center Photosystems (PS) PSI – oxygen from water splitting PSII – NADPH Proton Motive Force – ATP Photosynthesis The Electromagnetic Spectrum Photo-pigments 1 Photosynthesis – If Light Energy is Converted to Max Planck was a physicist who initiated the particle theory of light; its not just electromagnetic waves. Einstein ATP, how much light is needed? developed the concept of a photon, or a quanta of light. – What is the energy in ONE photon Thus, quantum mechanics was born. of light? Electromagnetic energy could be emitted only in quantized form, in other words, the energy could only be a – There is enough Light Energy to multiple of an elementary unit: E = h! ! = c/l make >5 ATP = h•c/l – How do you convert photons to where h is Planck's constant, also known as Planck's action quantum, and ! is the frequency of the radiation. Note that chemical energy? the units of energy are represented by h! and not simply by – You go through electrons…. !. So, the energy comes in units and not just a spectrum or – So we have to do re-dox (ox-phos continuum. in reverse! Energy of one mole of photons (1 einstein): 1928: Walther Nernst, Albert Einstein, Max Planck, Robert Millikan, –34 and Max Laue h = Planck’s constant (6.626 x 10 J•s) c = 3 x108 m/s l = 700 x 10–9 m E = 2.84 x -
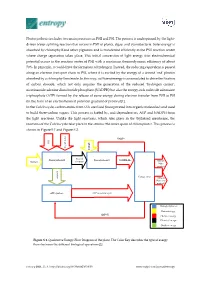
Photosynthesis Includes Two Main Processes As PSII and PSI
Photosynthesis includes two main processes as PSII and PSI. The process is underpinned by the light- driven water splitting reaction that occurs in PSII of plants, algae, and cyanobacteria. Solar energy is absorbed by chlorophyll and other pigments and is transferred efficiently to the PSII reaction center where charge separation takes place. This initial conversion of light energy into electrochemical potential occurs in the reaction center of PSII with a maximum thermodynamic efficiency of about 70%. In principle, it could drive the formation of hydrogen. Instead, the reducing equivalent is passed along an electron transport chain to PSI, where it is excited by the energy of a second ‘red’ photon absorbed by a chlorophyll molecule. In this way, sufficient energy is accumulated to drive the fixation of carbon dioxide, which not only requires the generation of the reduced ‘hydrogen carrier’, nicotinamide adenine dinucleotide phosphate (NADPH) but also the energy-rich molecule adenosine triphosphate (ATP) formed by the release of some energy during electron transfer from PSII to PSI (in the form of an electrochemical potential gradient of protons)[1]. In the Calvin cycle, carbon atoms from CO2 are fixed (incorporated into organic molecules) and used to build three-carbon sugars. This process is fueled by, and dependent on, ATP and NADPH from the light reactions. Unlike the light reactions, which take place in the thylakoid membrane, the reactions of the Calvin cycle take place in the stroma (the inner space of chloroplasts). This process is shown in Figure S 1 and Figure S 2. NADP+ CO2 Liquid water Excited Photosynthesis II Photosynthesis I NADPH+H+ Sunlight electrons Glucose Main product protons Calvine cycle Oxygen Waste(co- product) ATP synthase Biological process Photon Exergy ADP+Pi Chemical exergy Electrical exergy Gradient exergy Figure S 1. -

The Far-Red Limit of Photosynthesis
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1108 The Far-Red Limit of Photosynthesis FREDRIK MOKVIST ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-554-8835-2 UPPSALA urn:nbn:se:uu:diva-213659 2014 Dissertation presented at Uppsala University to be publicly examined in Siegbahnsalen, Ångströmlaboratoriet, Lägerhyddsvägen 1, Uppsala, Friday, 7 February 2014 at 10:15 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Professor Tomáš Polívka (University of South Bohemia in České Budějovice). Abstract Mokvist, F. 2014. The Far-Red Limit of Photosynthesis. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1108. 77 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-8835-2. The photosynthetic process has the unique ability to capture energy from sunlight and accumulate that energy in sugars and starch. This thesis deals with the light driven part of photosynthesis. The aim has been to investigate how the light-absorbing protein complexes Photosystem I (PS I) and Photosystem II (PS II), react upon illumination of light with lower energy (far-red light; 700-850 nm) than the absorption peak at respective primary donor, P700 and P680. The results were unexpected. At 295 K, we showed that both PS I and PS II were able to perform photochemistry with light up to 130 nm above its respective primary donor absorption maxima. As such, it was found that the primary donors’ action spectra extended approximately 80 nm further out into the red-region of the spectrum than previously reported. -

Photosystem II Secondary Article
Photosystem II Secondary article John Whitmarsh, University of Illinois, Urbana, Illinois, USA Article Contents Govindjee, University of Illinois, Urbana, Illinois, USA . Introduction . Organization, Composition and Structure Photosystem II is a specialized protein complex that uses light energy to oxidize water, . Light Capture: The Antenna System resulting in the release of molecular oxygen into the atmosphere, and to reduce . Primary Photochemistry: The Reaction Centre plastoquinone, which is released into the hydrophobic core of the photosynthetic . Oxidation of Water: The Source of Molecular Oxygen membrane. All oxygenic photosynthetic organisms, which include plants, algae and some . Reduction of Plastoquinone: The Two-electron Gate bacteria, depend on photosystem II to extract electrons from water that are eventually used . Photosystem II Contributes to a Proton Electrochemical Potential that Drives ATPase to reduce carbon dioxide in the carbon reduction cycle. The complex is composed of a . Downregulation: Energy is Diverted Away from the central reaction centre in which electron transport occurs, and a peripheral antenna system Reaction Centre when there is Excess Light which contains chlorophyll and other pigment molecules that absorb light. Secondary Electron Transfer Reactions in Photosystem II Protect Against Photodamage . Inactive Photosystem II: A Significant Proportion of Introduction Reaction Centres do not Work In Vivo . Fluorescence: Monitoring Photosystem II Activity In Photosynthetic organisms use light energy to produce Vivo organic molecules (Ort and Whitmarsh, 2001). In plants, . Summary algae and some types of bacteria, the photosynthetic process depends on photosystem II, a membrane-bound protein complexthat removes electrons from water and Chloroplasts originated from photosynthetic bacteria transfers them to plastoquinone, a specialized organic invading a nonphotosynthetic cell. -

The Complex Architecture of Oxygenic Photosynthesis
REVIEWS THE COMPLEX ARCHITECTURE OF OXYGENIC PHOTOSYNTHESIS Nathan Nelson and Adam Ben-Shem Abstract | Oxygenic photosynthesis is the principal producer of both oxygen and organic matter on earth. The primary step in this process — the conversion of sunlight into chemical energy — is driven by four, multisubunit, membrane-protein complexes that are known as photosystem I, photosystem II, cytochrome b6f and F-ATPase. Structural insights into these complexes are now providing a framework for the exploration not only of energy and electron transfer, but also of the evolutionary forces that shaped the photosynthetic apparatus. 8,9 PROTONMOTIVE FORCE Oxygenic photosynthesis — the conversion of sunlight After the invention of SDS–PAGE ,the biochemical (pmf). A special case of an into chemical energy by plants, green algae and composition of the four multisubunit protein com- electrochemical potential. It is cyanobacteria — underpins the survival of virtually all plexes began to be elucidated10–14.According to the par- the force that is created by the higher life forms. The production of oxygen and the tial reactions that they catalyse, PSII is defined as a accumulation of protons on one assimilation of carbon dioxide into organic matter water–plastoquinone oxidoreductase, the cytochrome-b f side of a cell membrane. This 6 concentration gradient is determines, to a large extent, the composition of our complex as a plastoquinone–plastocyanin oxidoreduc- generated using energy sources atmosphere and provides all life forms with essential tase, PSI as a plastocyanin–ferredoxin oxidoreductase such as redox potential or ATP. food and fuel. The study of the photosynthetic appara- and the F-ATPase as a pmf-driven ATP synthase15 (FIG. -

Determination of the Primary Charge Separation Rate in Isolated
Proc. Nati. Acad. Sci. USA Vol. 86, pp. 524-528, January 1989 Biophysics Determination of the primary charge separation rate in isolated photosystem II reaction centers with 500-fs time resolution (electron transfer/ultrafast spectroscopy/photosynthesis/Spinacia oleracea) MICHAEL R. WASIELEWSKIt, DOUGLAS G. JOHNSONt, MICHAEL SEIBERTt, AND GOVINDJEE§ tChemistry Division, Argonne National Laboratory, Argonne, IL 60439; tPhotoconversion Research Branch, Solar Energy Research Institute, Golden, CO 80401; and §Departments of Physiology and Biophysics, and Plant Biology, University of Illinois, Urbana, IL 61801 Communicated by Joseph J. Katz, October 3, 1988 ABSTRACT We have measured directly the rate of for- Pheoa - to a subsequent quinone electron acceptor takes mation of the oxidized chlorophyll a electron donor (P680') place in 510 ps. and the reduced electron acceptor pheophytin a- (Pheoa-) Until now it has proven very difficult to measure the following excitation ofisolated spinach photosystem II reaction primary charge separation rate in PSII RCs because isolation centers at 40C. The reaction-center complex consists of DI, D2, of a purified, stable RC preparation was not possible. Nanba and cytochrome b-559 proteins and was prepared by a proce- and Satoh (14) succeeded in isolating a complex that con- dure that stabilizes the protein complex. Transient absorption tained only D1, D2, and cytochrome b-559 proteins (see also difference spectra were measured from 440 to 850 nm as a refs. 15 and 16). Since this preparation bound four or five function of time with 500-fs resolution following 610-nm laser Chla molecules, two Pheoa molecules, and a single ,8- excitation. The formation of P680'-Pheoa- is indicated by the carotene molecule, and since Pheoa in the preparation was appearance of a band due to P680' at 820 nm and correspond- reduced following illumination, these authors suggested that ing absorbance changes at 505 and 540 nm due to formation of the complex was the RC ofPSII analogous to the RC ofpurple PheoaC. -

Light-Dependent Reactions of Photosynthesis
Light-dependent reactions of photosynthesis Tom Donald © 2015 American Society of Plant Biologists Lesson outline • Photosynthesis overview • Evolution and diversity of photosynthesis • Light and pigments • The light response curve and quantum efficiency • Plastids and chloroplasts • Structure and function of photosynthetic complexes • Pathways of electron transport • Damage avoidance and repair: Acclimations to light • Monitoring light reactions • Optimizing and improving photosynthesis • Artificial photosynthesis • Photosynthetic fungi and animals © 2015 American Society of Plant Biologists Overview: Photosynthesis captures light energy as reduced carbon “High energy”, Light-dependent reactions reduced The first step is the capture of carbon light energy as ATP and Energy input reducing power, NADPH from sunlight ATP NADPH Oxygen is Light-independent reactions released as The second step is the transfer of energy and reducing power from “Low energy”, a byproduct ATP and NADPH to CO , to produce oxidized carbon 2 high-energy, reduced sugars in carbon dioxide 6 CO2 + 6 H2O C6H12O6 + 6 O2 © 2015 American Society of Plant Biologists Photosynthesis is two sets of connected reactions 2 NADPH e− 2 H+ 2 NADP+ 2 H O O + 2 H+ + 2 e− ADP ATP Chloroplast 2 2 + H The LIGHT reactions take place in the thylakoid membranes The CARBON-FIXING reactions take place in the chloroplast stroma Adapted from Kramer, D.M., and Evans, J. R. (2010). The importance of energy balance in improving photosynthetic productivity. Plant Physiol.155: 70–78. © 2015 American Society of Plant Biologists Light reactions (usually) take place in thylakoid membranes Prokaryotes Eukaryotes Gloeobacter violaceus, Chlamydomonas the only cyanobacterium PLANT without thylakoid reinhardtii, a model green algae membranes Light-induced differentiation Proplastid Chloroplast Synechocystis spp.