1 SORBS2 Is a Susceptibility Gene to Arrhythmogenic Right Ventricular Cardiomyopathy Yonghe Ding, Phd,1, 2* Jingchun Yang, Phd 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

IDENTIFICATION and CHARACTERIZATION of ACTIN-REGULATORY PROTEINS in the HAIR CELL's CUTICULAR PLATE by LANA MARY POLLOCK Subm
IDENTIFICATION AND CHARACTERIZATION OF ACTIN-REGULATORY PROTEINS IN THE HAIR CELL’S CUTICULAR PLATE by LANA MARY POLLOCK Submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy Dissertation advisor: Brian M. McDermott Jr., Ph.D. Department of Genetics and Genome Sciences CASE WESTERN RESERVE UNIVERSITY January 2016 Case Western Reserve University School of Graduate Studies We, the thesis committee, hereby approve the thesis/dissertation of Lana Pollock, candidate for the degree of Doctor of Philosophy (PhD).* (signed)_________Zhenghe Wang, Ph.D._________________ (chair of committee) ___________Brian McDermott, Ph.D._______________ ___________ Hua Lou, Ph.D._____________________ ___________Stephen Maricich, Ph.D., M.D.___________ ___________Anthony Wynshaw-Boris, Ph.D., M.D._____ Date of defense_____September 8th, 2015_______________ *we also certify that written approval has been obtained for release of any proprietary material contained therein 2 This thesis is dedicated to Daniel Margevicius. Thank you for your unwavering love and support. Ačiū!! 3 Table of contents List of Tables ........................................................................................................ 7 List of Figures ....................................................................................................... 8 List of abbreviations ............................................................................................ 13 Abstract ............................................................................................................. -

Vinexin Family (SORBS) Proteins Play Different Roles in Stiffness- Sensing and Contractile Force Generation Takafumi Ichikawa1,2, Masahiro Kita1, Tsubasa S
© 2017. Published by The Company of Biologists Ltd | Journal of Cell Science (2017) 130, 3517-3531 doi:10.1242/jcs.200691 RESEARCH ARTICLE Vinexin family (SORBS) proteins play different roles in stiffness- sensing and contractile force generation Takafumi Ichikawa1,2, Masahiro Kita1, Tsubasa S. Matsui3,4, Ayaka Ichikawa Nagasato1, Tomohiko Araki3, Shian-Huey Chiang5, Takuhito Sezaki1, Yasuhisa Kimura1, Kazumitsu Ueda1,2, Shinji Deguchi3,4, Alan R. Saltiel5,* and Noriyuki Kioka1,2,‡ ABSTRACT generating actin stress fibers (SFs) (Geiger et al., 2001). This Vinexin, c-Cbl associated protein (CAP) and Arg-binding protein 2 ‘ ’ (ArgBP2) constitute an adaptor protein family called the vinexin mechanical linkage acts as a molecular clutch to transmit the force (SORBS) family that is targeted to focal adhesions (FAs). Although derived from non-muscle myosin-II-dependent contraction to the numerous studies have focused on each of the SORBS proteins and ECM. Cells on more rigid substrates exert greater contractile forces partially elucidated their involvement in mechanotransduction, a than those on soft substrates (Hoffman et al., 2011; Roca-Cusachs comparative analysis of their function has not been well addressed. et al., 2012; LaCroix et al., 2015). These alterations can lead to Here, we established mouse embryonic fibroblasts that individually stiffness-dependent biochemical signals. Among the numerous FA scaffolding proteins, vinculin is one of expressed SORBS proteins and analysed their functions in an ‘ ’ identical cell context. Both vinexin-α and CAP co-localized with the main clutch molecules that can regulate force transmission. vinculin at FAs and promoted the appearance of vinculin-rich FAs, Vinculin consists of an N-terminal head region and a C-terminal tail α region separated by a flexible proline-rich linker region (Bakolitsa whereas ArgBP2 co-localized with -actinin at the proximal end of – FAs and punctate structures on actin stress fibers (SFs), and induced et al., 2004; Borgon et al., 2004). -

SORBS2 Transcription Is Activated by Telomere Position Effect–Over Long Distance Upon Telomere Shortening in Muscle Cells From
SORBS2 transcription is activated by telomere position effect–over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy Jérôme Robin, Andrew Ludlow, Kimberly Batten, Marie-Cécile Gaillard, Guido Stadler, Frédérique Magdinier, Woodring Wright, Jerry W. Shay To cite this version: Jérôme Robin, Andrew Ludlow, Kimberly Batten, Marie-Cécile Gaillard, Guido Stadler, et al.. SORBS2 transcription is activated by telomere position effect–over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy. Genome Research, Cold Spring Harbor Laboratory Press, 2015, 25 (12), pp.1781 - 1790. 10.1101/gr.190660.115. hal- 01663663 HAL Id: hal-01663663 https://hal-amu.archives-ouvertes.fr/hal-01663663 Submitted on 14 Dec 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Downloaded from genome.cshlp.org on December 13, 2017 - Published by Cold Spring Harbor Laboratory Press Research SORBS2 transcription is activated by telomere position effect–over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy Jérôme D. Robin,1 Andrew T. Ludlow,1 Kimberly Batten,1 Marie-Cécile Gaillard,2 Guido Stadler,1 Frédérique Magdinier,2 Woodring E. -

Phosphoproteomic Profiling of Human Myocardial Tissues Distinguishes Ischemic from Non-Ischemic End Stage Heart Failure
Phosphoproteomic Profiling of Human Myocardial Tissues Distinguishes Ischemic from Non-Ischemic End Stage Heart Failure Matthew A. Schechter1., Michael K. H. Hsieh1., Linda W. Njoroge1, J. Will Thompson2, Erik J. Soderblom2, Bryan J. Feger1, Constantine D. Troupes1, Kathleen A. Hershberger3,4, Olga R. Ilkayeva3, Whitney L. Nagel1, Gina P. Landinez1, Kishan M. Shah1, Virginia A. Burns5, Lucia Santacruz1, Matthew D. Hirschey3,4, Matthew W. Foster6, Carmelo A. Milano1, M. Arthur Moseley2, Valentino Piacentino III1, Dawn E. Bowles1* 1 Department of Surgery, Duke University Medical Center, Durham, North Carolina, United States of America, 2 Duke Proteomics Core, Duke University Medical Center, Durham, North Carolina, United States of America, 3 Sarah W. Stedman Nutrition and Metabolism Center, Duke University Medical Center, Durham, North Carolina, United States of America, 4 Department of Medicine, Duke University Medical Center, Durham, North Carolina, United States of America, 5 Duke Translational Research Institute, Duke University Medical Center, Durham, North Carolina, United States of America, 6 Division of Pulmonary, Allergy and Critical Care, Medicine, Duke University Medical Center, Durham, North Carolina, United States of America Abstract The molecular differences between ischemic (IF) and non-ischemic (NIF) heart failure are poorly defined. A better understanding of the molecular differences between these two heart failure etiologies may lead to the development of more effective heart failure therapeutics. In this study extensive -

SORBS2 Transcription Is Activated by Telomere Position Effect–Over Long
Downloaded from genome.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press Research SORBS2 transcription is activated by telomere position effect–over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy Jérôme D. Robin,1 Andrew T. Ludlow,1 Kimberly Batten,1 Marie-Cécile Gaillard,2 Guido Stadler,1 Frédérique Magdinier,2 Woodring E. Wright,1 and Jerry W. Shay1,3 1Department of Cell Biology, UT Southwestern Medical Center, Dallas, Texas 75390, USA; 2Aix Marseille Universite, INSERM, GMGF, Marseille 13385 Cedex 05, France; 3Center for Excellence in Genomics Medicine Research, King Abdulaziz University, Jeddah 21589, Saudi Arabia DNA is organized into complex three-dimensional chromatin structures, but how this spatial organization regulates gene expression remains a central question. These DNA/chromatin looping structures can range in size from 10–20 kb (enhanc- ers/repressors) to many megabases during intra- and inter-chromosomal interactions. Recently, the influence of telomere length on chromatin organization prior to senescence has revealed the existence of long-distance chromatin loops that dic- tate the expression of genes located up to 10 Mb from the telomeres (Telomere Position Effect–Over Long Distances [TPE- OLD]). Here, we demonstrate the existence of a telomere loop at the 4q35 locus involving the sorbin and SH3 domain-con- taining protein 2 gene, SORBS2, a skeletal muscle protein using a modification of the chromosome conformation capture method. The loop reveals a cis-acting mechanism modifying SORBS2 transcription. The expression of this gene is altered by TPE-OLD in myoblasts from patients affected with the age-associated genetic disease, facioscapulohumeral muscular dys- trophy (FSHD1A, MIM 158900). -

Phenotype Informatics
Freie Universit¨atBerlin Department of Mathematics and Computer Science Phenotype informatics: Network approaches towards understanding the diseasome Sebastian Kohler¨ Submitted on: 12th September 2012 Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) am Fachbereich Mathematik und Informatik der Freien Universitat¨ Berlin ii 1. Gutachter Prof. Dr. Martin Vingron 2. Gutachter: Prof. Dr. Peter N. Robinson 3. Gutachter: Christopher J. Mungall, Ph.D. Tag der Disputation: 16.05.2013 Preface This thesis presents research work on novel computational approaches to investigate and characterise the association between genes and pheno- typic abnormalities. It demonstrates methods for organisation, integra- tion, and mining of phenotype data in the field of genetics, with special application to human genetics. Here I will describe the parts of this the- sis that have been published in peer-reviewed journals. Often in modern science different people from different institutions contribute to research projects. The same is true for this thesis, and thus I will itemise who was responsible for specific sub-projects. In chapter 2, a new method for associating genes to phenotypes by means of protein-protein-interaction networks is described. I present a strategy to organise disease data and show how this can be used to link diseases to the corresponding genes. I show that global network distance measure in interaction networks of proteins is well suited for investigat- ing genotype-phenotype associations. This work has been published in 2008 in the American Journal of Human Genetics. My contribution here was to plan the project, implement the software, and finally test and evaluate the method on human genetics data; the implementation part was done in close collaboration with Sebastian Bauer. -

SORBS2 Transcription Is Activated by Telomere Position Effect-Over Long
Downloaded from genome.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press SORBS2 Transcription is Activated by Telomere Position Effect-Over Long Distance Upon Telomere Shortening in Muscle Cells from Patients with Facioscapulohumeral Dystrophy Jérôme D. Robin1,4 Andrew T. Ludlow1 Kimberly Batten1 Marie-Cecile Gaillard2; Guido Stadler1; Frédérique Magdinier2; Woodring E. Wright1; Jerry W. Shay1,3,4 1 Department of Cell Biology, UT Southwestern Medical Center, Dallas TX 75390 U.S.A. 2 Aix Marseille Universite, INSERM, GMGF, UMRS 910. 27 Bd Jean Moulin, Marseille 13385 Cedex 05 France. 3 Center for Excellence in Genomics Medicine Research, King Abdulaziz University, Jeddah, Saudi Arabia 4 Co-Corresponding Authors: Jerry W. Shay Tel: 214-648-4201 Fax: 214-648-5814 [email protected] Jérôme D. Robin IRCAN CNRS UMR 7284 / INSERM U1081 Faculté de Médecine 28 avenue de Valombrose 06107 Nice Cedex 2 [email protected] Keywords: chromatin, 4q35 locus, D4Z4, skeletal muscle, myoblast, Hi-C, Telomere, Facioscapulohumeral Dystrophy Downloaded from genome.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract DNA is organized into complex three-dimensional chromatin structures but how this special organization regulates gene expression remains a central question. These DNA/chromatin looping structures can range in size from 10-20 kilobases (enhancers/repressors) to many megabases (Mb) during intra- and inter- chromosomal interactions. Recently, the influence of telomere length on chromatin organization prior to senescence has revealed the existence of long distance chromatin loops that dictate the expression of genes located up to 10Mb from the telomeres (Telomere Position Effect-Over Long Distances; TPE-OLD). -

Range Interactions in Patients Affected with Facio-Scapulo
www.nature.com/scientificreports OPEN Analysis of the 4q35 chromatin organization reveals distinct long- range interactions in patients Received: 13 March 2019 Accepted: 25 June 2019 afected with Facio-Scapulo- Published: xx xx xxxx Humeral Dystrophy Marie-Cécile Gaillard1, Natacha Broucqsault1, Julia Morere1, Camille Laberthonnière1, Camille Dion1, Cherif Badja1, Stéphane Roche1, Karine Nguyen1,2, Frédérique Magdinier1 & Jérôme D. Robin 1 Facio-Scapulo Humeral dystrophy (FSHD) is the third most common myopathy, afecting 1 amongst 10,000 individuals (FSHD1, OMIM #158900). This autosomal dominant pathology is associated in 95% of cases with genetic and epigenetic alterations in the subtelomeric region at the extremity of the long arm of chromosome 4 (q arm). A large proportion of the remaining 5% of cases carry a mutation in the SMCHD1 gene (FSHD2, OMIM #158901). Here, we explored the 3D organization of the 4q35 locus by three-dimensions DNA in situ fuorescent hybridization (3D-FISH) in primary fbroblasts isolated from patients and healthy donors. We found that D4Z4 contractions and/or SMCHD1 mutations impact the spatial organization of the 4q35 region and trigger changes in the expression of diferent genes. Changes in gene expression were corroborated in muscle biopsies suggesting that the modifed chromatin landscape impelled a modulation in the level of expression of a number of genes across the 4q35 locus in FSHD. Using induced pluripotent stem cells (hIPSC), we further examined whether chromatin organization is inherited after reprogramming or acquired during diferentiation and showed that folding of the 4q35 region is modifed upon diferentiation. These results together with previous fndings highlight the role of the D4Z4 macrosatellite repeat in the topological organization of chromatin and further indicate that the D4Z4-dependent 3D structure induces transcriptional changes of 4q35 genes expression. -

Terminal Chromosome 4Q Deletion Syndrome in an Infant with Hearing
Vona et al. BMC Medical Genetics 2014, 15:72 http://www.biomedcentral.com/1471-2350/15/72 CASE REPORT Open Access Terminal chromosome 4q deletion syndrome in an infant with hearing impairment and moderate syndromic features: review of literature Barbara Vona1, Indrajit Nanda1, Cordula Neuner1, Jörg Schröder1, Vera M Kalscheuer2, Wafaa Shehata-Dieler3 and Thomas Haaf1* Abstract Background: Terminal deletions of chromosome 4q are associated with a broad spectrum of phenotypes including cardiac, craniofacial, digital, and cognitive impairment. The rarity of this syndrome renders genotype-phenotype correlation difficult, which is further complicated by the widely different phenotypes observed in patients sharing similar deletion intervals. Case presentation: Herein, we describe a boy with congenital hearing impairment and a variety of moderate syndromic features that prompted SNP array analysis disclosing a heterozygous 6.9 Mb deletion in the 4q35.1q35.2 region, which emerged de novo in the maternal germ line. Conclusion: In addition to the index patient, we review 35 cases from the literature and DECIPHER database to attempt genotype-phenotype correlations for a syndrome with great phenotypic variability. We delineate intervals with recurrent phenotypic overlap, particularly for cleft palate, congenital heart defect, intellectual disability, and autism spectrum disorder. Broad phenotypic presentation of the terminal 4q deletion syndrome is consistent with incomplete penetrance of the individual symptoms. Keywords: Genotype-phenotype association, Copy number variation, Parent-of-origin, SNP array, Terminal 4q deletion syndrome Background skeletal and digital abnormalities, and occasionally autism Terminal deletions of chromosome 4q are a rare event spectrum disorder (ASD), behavioural disorders, and de- with an approximate incidence of 1 in 100,000 [1,2]. -
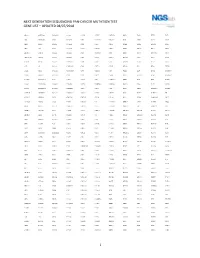
1 Next Generation Sequencing Pan-Cancer Mutation Test Gene List – Updated 08/07/2018
NEXT GENERATION SEQUENCING PAN-CANCER MUTATION TEST GENE LIST – UPDATED 08/07/2018 ABCC3 ANKRD26 BAIAP2L1 C11orf1 CCT6B CENPU CREB3L2 DDX6 EGR2 ETV5 FGF3 ABI1 ANKRD28 BAP1 C11orf30 CD19 CEP170B CREBBP DEK EGR3 ETV6 FGF4 ABL1 ANLN BARD1 C11orf54 CD22 CEP57 CRKL DGKB EGR4 EWSR1 FGF5 ABL2 APC BAX C11orf95 CD274 CEP85L CRLF2 DGKI EIF4A2 EXO1 FGF6 ABLIM1 APH1A BAZ2A C2CD2L CD28 CHCHD7 CRTC1 DGKZ EIF4E EXOSC6 FGF7 ABRAXAS1 APLP2 BCAS3 C2orf44 CD36 CHD2 CRTC3 DICER1 ELF4 EXT1 FGF8 ACACA APOD BCAS4 CACNA1F CD44 CHD6 CSF1 DIRAS3 ELK4 EXT2 FGF9 ACE AR BCL10 CACNA1G CD58 CHEK1 CSF1R DIS3L2 ELL EYA1 FGFR1 ACER1 ARAF BCL11A CACNA2D3 CD70 CHEK2 CSF3 DKK1 ELN EYA2 FGFR1OP ACKR3 ARFRP1 BCL11B CAD CD74 CHIC2 CSF3R DKK2 ELOVL2 EZH2 FGFR1OP2 ACSBG1 ARHGAP20 BCL2 CALR CD79A CHL1 CSNK1G2 DKK4 ELP2 EZR FGFR2 ACSL3 ARHGAP26 BCL2A1 CAMK2A CD79B CHMP2B CSNK2A1 DLEC1 EML1 FAF1 FGFR3 ACSL6 ARHGEF12 BCL2L1 CAMK2B CD8A CHN1 CTCF DLL1 EML4 FAM127C FGFR4 ACVR1B ARHGEF7 BCL2L2 CAMK2G CDC14A CHST11 CTDSP2 DLL3 ENPP2 FAM19A2 FH ACVR1C ARID1A BCL3 CAMTA1 CDC14B CHUK CTLA4 DLL4 EP300 FAM19A5 FHIT ACVR2A ARID2 BCL6 CANT1 CDC25A CIC CTNNA1 DMRT1 EP400 FAM46C FHL2 ADD3 ARIH2 BCL7A CAPRIN1 CDC25C CIITA CTNNB1 DMRTA2 EPC1 FAM64A FIGF ADGRA2 ARL6IP5 BCL9 CAPZB CDC42 CIRH1A CTNND2 DNAJB1 EPCAM FANCA FIP1L1 ADGRG7 ARNT BCOR CARD11 CDC73 CIT CTRB1 DNM1 EPHA10 FANCB FLCN ADM ARRDC4 BCORL1 CARM1 CDH1 CKB CTSA DNM2 EPHA2 FANCC FLI1 AFF1 ASMTL BCR CARS CDH11 CKS1B CUL4A DNM3 EPHA3 FANCD2 FLNA AFF3 ASPH BDNF CASC5 CDK1 CLP1 CUL4B DNMT1 EPHA5 FANCE FLNC AFF4 ASPSCR1 BHLHE22 CASP3 CDK12 -

Nucleolin Is Essential for Rabbit Hemorrhagic Disease Virus Replication By
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.13.094185; this version posted May 13, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 Nucleolin is essential for rabbit hemorrhagic disease virus replication by 3 providing a physical link in replication complex formation 4 5 Jie Zhu1, Qiuhong Miao1, 2, Hongyuan Guo1, Ruibin Qi1, Aoxing Tang1, Dandan Dong1, 6 Jingyu Tang1, Guangzhi Tong1*, Guangqing Liu1* 7 8 1 Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences, 9 Shanghai, 200241, China; 10 2 Laboratory of Virology, Wageningen University and Research, Wageningen, 6708 PB, The 11 Netherlands; 12 13 *Corresponding author 14 E-mail: [email protected] (GL); [email protected] (GT) 15 16 17 18 19 20 21 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.13.094185; this version posted May 13, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 22 Abstract 23 Rabbit hemorrhagic disease virus (RHDV) is an important member of the Caliciviridae 24 family and cannot be propagated in vitro, which has greatly impeded progress of 25 investigating its replication mechanism. Construction of an RHDV replicon system has 26 recently provided a platform for exploring RHDV replication in host cells. -
Alterations of Actin Cytoskeleton and Arterial Protein Amounts in Patients with Obstructive Jaundice
Alterations of actin cytoskeleton and arterial protein amounts in patients with obstructive jaundice. Hongqian Wang second military medical university Xiaoyan Meng second military medical university Mo Chen second military medical university Jinming Zhang second military medical university Baohua Zhang second military medical university Feixiang Wu ( [email protected] ) second military medical university, eastern hepatobiliary surgical hospital. https://orcid.org/0000- 0003-1149-3894 Research article Keywords: Vascular hypo-reactivity; obstructive jaundice; proteomics; artery; hemodynamic changes. Posted Date: June 27th, 2019 DOI: https://doi.org/10.21203/rs.2.10394/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/16 Abstract Background: Vascular hypo-responsiveness to vasopressors in patients with obstructive jaundice (OJ) is a common anesthetic event, which leads to perioperative complications and increased mortality. The cause of this clinical issue remains unclear. Here, changes in the artery of OJ patients were assessed by proteomic analysis. Methods: Ten patients with OJ due to bile duct or pancreatic head neoplasms were enrolled, alongside ten chronic cholecystitis or liver hemangioma cases forming the controls group. Vascular reactivity was measured before anesthesia on the day of surgery. Artery samples near or surrounding the removed tumor tissues were collected and evaluated by 2-dimensional electrophoresis. Proteins with differential expression were detected by MALDI-TOF mass spectrometry, with immunoblot conrmation. Results: Vascular hypo-reactivity in OJ cases as well as suppressed aortic response to vasoactive products were evidenced. We also found that actin cytoskeleton and several actin-binding proteins were up- or down-regulated in the artery of OJ patients.