Cysteinyl Leukotriene Receptor 1/2 Antagonists Nonselectively Modulate Organic Anion Transport by Multidrug Resistance Proteins (MRP1-4) S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 5-HT6 Receptor Antagonist SB-271046 Selectively Enhances Excitatory Neurotransmission in the Rat Frontal Cortex and Hippocampus Lee A
The 5-HT6 Receptor Antagonist SB-271046 Selectively Enhances Excitatory Neurotransmission in the Rat Frontal Cortex and Hippocampus Lee A. Dawson, Ph.D., Huy Q. Nguyen, B.S., and Ping Li, B.S. Preclinical evidence has suggested a possible role for the 5-HT6 increases in extracellular glutamate levels in both frontal receptor in the treatment of cognitive dysfunction. However, cortex and dorsal hippocampus, respectively. These effects were currently there is little neurochemical evidence suggesting the completely attenuated by infusion of tetrodotoxin but mechanism(s) which may be involved. Using the selective unaffected by the muscarinic antagonist, atropine. Here we 5-HT6 antagonist SB-271046 and in vivo microdialysis, we demonstrate for the first time the selective enhancement of have evaluated the effects of this compound on the modulation excitatory neurotransmission by SB-271046 in those brain of basal neurotransmitter release within multiple brain regions regions implicated in cognitive and memory function, and of the freely moving rat. SB-271046 produced no change in provide mechanistic evidence in support of a possible basal levels of dopamine (DA), norepinephrine (NE) or 5-HT therapeutic role for 5-HT6 receptor antagonists in the in the striatum, frontal cortex, dorsal hippocampus or nucleus treatment of cognitive and memory dysfunction. accumbens. Similarly, this compound had no effect on [Neuropsychopharmacology 25:662–668, 2001] excitatory neurotransmission in the striatum or nucleus © 2001 American College of Neuropsychopharmacology. accumbens. Conversely, SB-271046 produced 3- and 2-fold Published by Elsevier Science Inc. KEY WORDS: 5-HT6 receptor; SB-271046; Microdialysis; sma et al. 1993; Ruat et al. -

Receptor Antagonist (H RA) Shortages | May 25, 2020 2 2 2 GERD4,5 • Take This Opportunity to Determine If Continued Treatment Is Necessary
H2-receptor antagonist (H2RA) Shortages Background . 2 H2RA Alternatives . 2 Therapeutic Alternatives . 2 Adults . 2 GERD . 3 PUD . 3 Pediatrics . 3 GERD . 3 PUD . 4 Tables Table 1: Health Canada–Approved Indications of H2RAs . 2 Table 2: Oral Adult Doses of H2RAs and PPIs for GERD . 4 Table 3: Oral Adult Doses of H2RAs and PPIs for PUD . 5 Table 4: Oral Pediatric Doses of H2RAs and PPIs for GERD . 6 Table 5: Oral Pediatric Doses of H2RAs and PPIs for PUD . 7 References . 8 H2-receptor antagonist (H2RA) Shortages | May 25, 2020 1 H2-receptor antagonist (H2RA) Shortages BACKGROUND Health Canada recalls1 and manufacturer supply disruptions may be causing shortages of commonly used acid-reducing medications called histamine H2-receptor antagonists (H2RAs) . H2RAs include cimetidine, famotidine, nizatidine and ranitidine . 2 There are several Health Canada–approved indications of H2RAs (see Table 1); this document addresses the most common: gastroesophageal reflux disease (GERD) and peptic ulcer disease (PUD) . 2 TABLE 1: HEALTH CANADA–APPROVED INDICATIONS OF H2RAs H -Receptor Antagonists (H RAs) Health Canada–Approved Indications 2 2 Cimetidine Famotidine Nizatidine Ranitidine Duodenal ulcer, treatment ü ü ü ü Duodenal ulcer, prophylaxis — ü ü ü Benign gastric ulcer, treatment ü ü ü ü Gastric ulcer, prophylaxis — — — ü GERD, treatment ü ü ü ü GERD, maintenance of remission — ü — — Gastric hypersecretion,* treatment ü ü — ü Self-medication of acid indigestion, treatment and prophylaxis — ü† — ü† Acid aspiration syndrome, prophylaxis — — — ü Hemorrhage from stress ulceration or recurrent bleeding, — — — ü prophylaxis ü = Health Canada–approved indication; GERD = gastroesophageal reflux disease *For example, Zollinger-Ellison syndrome . -

Binding Mode Exploration of B1 Receptor Antagonists' by the Use of Molecular Dynamics and Docking Simulation—How Different T
International Journal of Molecular Sciences Article Binding Mode Exploration of B1 Receptor Antagonists’ by the Use of Molecular Dynamics and Docking Simulation—How Different Target Engagement Can Determine Different Biological Effects Marica Gemei 1,*, Carmine Talarico 1 , Laura Brandolini 1, Candida Manelfi 1, Lorena Za 2, Silvia Bovolenta 2, Chiara Liberati 2, Luigi Del Vecchio 3, Roberto Russo 4 , Carmen Cerchia 4, Marcello Allegretti 1 and Andrea Rosario Beccari 1 1 Dompé Farmaceutici SpA, via Campo di Pile, 67100 L’Aquila, Italy; [email protected] (C.T.); [email protected] (L.B.); candida.manelfi@dompe.com (C.M.); [email protected] (M.A.); [email protected] (A.R.B.) 2 Axxam, Via Meucci 3, Bresso, 20091 Milano, Italy; [email protected] (L.Z.); [email protected] (S.B.); [email protected] (C.L.) 3 Ceinge Biotecnologie Avanzate, via G. Salvatore 486, 80145 Napoli, Italy; [email protected] 4 Department of Pharmacy, University of Naples “Federico II”, via D. Montesano, 49, 80131 Napoli, Italy; [email protected] (R.R.); [email protected] (C.C.) * Correspondence: [email protected]; Tel.: +34-06-465916 Received: 26 August 2020; Accepted: 12 October 2020; Published: 16 October 2020 Abstract: The kinin B1 receptor plays a critical role in the chronic phase of pain and inflammation. The development of B1 antagonists peaked in recent years but almost all promising molecules failed in clinical trials. Little is known about these molecules’ mechanisms of action and additional information will be necessary to exploit the potential of the B1 receptor. -

The Selective Serotonin2a Receptor Antagonist, MDL100,907, Elicits A
BRIEF REPORT The Selective Serotonin2A Receptor Antagonist, MDL100,907, Elicits a Specific Interoceptive Cue in Rats Anne Dekeyne, Ph.D., Loretta Iob, B.Sc., Patrick Hautefaye, Ph.D., and Mark J. Millan, Ph.D. Employing a two-lever, food-reinforced, Fixed Ratio 10 5-HT2B/2C antagonist, SB206,553 (0.16 and 2.5 mg/kg) and drug discrimination procedure, rats were trained to the selective 5-HT2C antagonists, SB242,084 (2.5 and recognize the highly-selective serotonin (5-HT)2A receptor 10.0 mg/kg,) and RS102221 (2.5 and 10.0 mg/kg), did not antagonist, MDL100,907 (0.16 mg/kg, i.p.). They attained significantly generalize. In conclusion, selective blockade of Ϯ Ϯ criterion after a mean S.E.M. of 70 11 sessions. 5-HT2A receptors by MDL100,907 elicits a discriminative MDL100,907 fully generalized with an Effective Dose stimulus in rats which appears to be specifically mediated (ED)50 of 0.005 mg/kg, s.c.. A further selective 5-HT2A via 5-HT2A as compared with 5-HT2B and 5-HT2C receptors. antagonist, SR46349, similarly generalized with an ED50 of [Neuropsychopharmacology 26:552–556, 2002] 0.04 mg/kg, s.c. In distinction, the selective 5-HT2B © 2002 American College of Neuropsychopharmacology antagonist, SB204,741 (0.63 and 10.0 mg/kg), the Published by Elsevier Science Inc. KEY WORDS: Drug discrimination; Interoceptive; 5-HT2A stimulus (DS) properties of several 5-HT2 agonists and receptors hallucinogens, such as mescaline (Appel and Callahan 1989), lysergic acid diethylamide (LSD) (Fiorella et al. Drug discrimination procedures have been extensively 1995) and quipazine (Friedman et al. -

Pharmacology/Therapeutics II Block I Lectures – 2013‐14
Pharmacology/Therapeutics II Block I Lectures – 2013‐14 54. H2 Blocker, PPls – Patel 55. Principles of Clinical Toxicology – Kennedy 56. Anti‐Parasitic Agents – Johnson (To be posted later) 57. Palliation of Constipation & Nausea/vomiting – Kristopaitis (Lecture in Room 190) Tarun B. Patel, Ph.D Date: January 9, 2013: 10:30 a.m. Reading Assignment: Katzung, Basic and Clinical Pharmacology, 11th Edition, pp. 1067-1077. KEY CONCEPTS AND LEARNING OBJECTIVES Histamine via its different receptors produces a number of physiological and pathological actions. Therefore, anti-histaminergic drugs may be used to treat different conditions. 1. To know the physiological functions of histamine. 2. To understand which histamine receptors mediate the different effects of histamine in stomach ulcers. 3. To know what stimuli cause the release of histamine and acid in stomach. 4. To know the types of histamine H2 receptor antagonists that are available clinically. 5. To know the clinical uses of H2 receptor antagonists. 6. To know the drug interactions associated with the use of H2 receptor antagonists. 7. To understand the mechanism of action of PPIs 8. To know the adverse effects and drugs interactions with PPIs 9. To know the role of H. pylori in gastric ulceration 10. To know the drugs used to treat H. pylori infection Drug List: See Summary Table Provided at end of handout. Page 1 Tarun B. Patel, Ph.D Histamine H2 receptor antagonists and PPIs in the treatment of GI Ulcers: The following section covers medicines used to treat ulcer. These medicines include H2 receptor antagonists, proton pump inhibitors, mucosal protective agents and antibiotics (for treatment of H. -

Drug Action-Receptor Theory
Drug action-Receptor Theory Molecules (eg, drugs, hormones, neurotransmitters) that bind to a receptor are called ligands. The binding can be specific and reversible. A ligand may activate or inactivate a receptor; activation may increase or decrease a particular cell function. Each ligand may interact with multiple receptor subtypes. Few if any drugs are absolutely specific for one receptor or subtype, but most have relative selectivity. Selectivity is the degree to which a drug acts on a given site relative to other sites; selectivity relates largely to physicochemical binding of the drug to cellular receptors. A drug’s ability to affect a given receptor is related to the drug’s affinity (probability of the drug occupying a receptor at any given instant) and intrinsic efficacy (intrinsic activity—degree to which a ligand activates receptors and leads to cellular response). A drug’s affinity and activity are determined by its chemical structure. The pharmacologic effect is also determined by the duration of time that the drug-receptor complex persists (residence time). The lifetime of the drug-receptor complex is affected by dynamic processes (conformation changes) that control the rate of drug association and dissociation from the target. A longer residence time explains a prolonged pharmacologic effect. Drugs with long residence times include finasteride and darunavir. A longer residence time can be a potential disadvantage when it prolongs a drug's toxicity. For some receptors, transient drug occupancy produces the desired pharmacologic effect, whereas prolonged occupancy causes toxicity. Ability to bind to a receptor is influenced by external factors as well as by intracellular regulatory mechanisms. -

The Significance of NK1 Receptor Ligands and Their Application In
pharmaceutics Review The Significance of NK1 Receptor Ligands and Their Application in Targeted Radionuclide Tumour Therapy Agnieszka Majkowska-Pilip * , Paweł Krzysztof Halik and Ewa Gniazdowska Centre of Radiochemistry and Nuclear Chemistry, Institute of Nuclear Chemistry and Technology, Dorodna 16, 03-195 Warsaw, Poland * Correspondence: [email protected]; Tel.: +48-22-504-10-11 Received: 7 June 2019; Accepted: 16 August 2019; Published: 1 September 2019 Abstract: To date, our understanding of the Substance P (SP) and neurokinin 1 receptor (NK1R) system shows intricate relations between human physiology and disease occurrence or progression. Within the oncological field, overexpression of NK1R and this SP/NK1R system have been implicated in cancer cell progression and poor overall prognosis. This review focuses on providing an update on the current state of knowledge around the wide spectrum of NK1R ligands and applications of radioligands as radiopharmaceuticals. In this review, data concerning both the chemical and biological aspects of peptide and nonpeptide ligands as agonists or antagonists in classical and nuclear medicine, are presented and discussed. However, the research presented here is primarily focused on NK1R nonpeptide antagonistic ligands and the potential application of SP/NK1R system in targeted radionuclide tumour therapy. Keywords: neurokinin 1 receptor; Substance P; SP analogues; NK1R antagonists; targeted therapy; radioligands; tumour therapy; PET imaging 1. Introduction Neurokinin 1 receptor (NK1R), also known as tachykinin receptor 1 (TACR1), belongs to the tachykinin receptor subfamily of G protein-coupled receptors (GPCRs), also called seven-transmembrane domain receptors (Figure1)[ 1–3]. The human NK1 receptor structure [4] is available in Protein Data Bank (6E59). -

Pharmacokinetics and Pharmacology of Drugs Used in Children
Drug and Fluid Th erapy SECTION II Pharmacokinetics and Pharmacology of Drugs Used CHAPTER 6 in Children Charles J. Coté, Jerrold Lerman, Robert M. Ward, Ralph A. Lugo, and Nishan Goudsouzian Drug Distribution Propofol Protein Binding Ketamine Body Composition Etomidate Metabolism and Excretion Muscle Relaxants Hepatic Blood Flow Succinylcholine Renal Excretion Intermediate-Acting Nondepolarizing Relaxants Pharmacokinetic Principles and Calculations Atracurium First-Order Kinetics Cisatracurium Half-Life Vecuronium First-Order Single-Compartment Kinetics Rocuronium First-Order Multiple-Compartment Kinetics Clinical Implications When Using Short- and Zero-Order Kinetics Intermediate-Acting Relaxants Apparent Volume of Distribution Long-Acting Nondepolarizing Relaxants Repetitive Dosing and Drug Accumulation Pancuronium Steady State Antagonism of Muscle Relaxants Loading Dose General Principles Central Nervous System Effects Suggamadex The Drug Approval Process, the Package Insert, and Relaxants in Special Situations Drug Labeling Opioids Inhalation Anesthetic Agents Morphine Physicochemical Properties Meperidine Pharmacokinetics of Inhaled Anesthetics Hydromorphone Pharmacodynamics of Inhaled Anesthetics Oxycodone Clinical Effects Methadone Nitrous Oxide Fentanyl Environmental Impact Alfentanil Oxygen Sufentanil Intravenous Anesthetic Agents Remifentanil Barbiturates Butorphanol and Nalbuphine 89 A Practice of Anesthesia for Infants and Children Codeine Antiemetics Tramadol Metoclopramide Nonsteroidal Anti-infl ammatory Agents 5-Hydroxytryptamine -

Anew Drug Design Strategy in the Liht of Molecular Hybridization Concept
www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 “Drug Design strategy and chemical process maximization in the light of Molecular Hybridization Concept.” Subhasis Basu, Ph D Registration No: VB 1198 of 2018-2019. Department Of Chemistry, Visva-Bharati University A Draft Thesis is submitted for the partial fulfilment of PhD in Chemistry Thesis/Degree proceeding. DECLARATION I Certify that a. The Work contained in this thesis is original and has been done by me under the guidance of my supervisor. b. The work has not been submitted to any other Institute for any degree or diploma. c. I have followed the guidelines provided by the Institute in preparing the thesis. d. I have conformed to the norms and guidelines given in the Ethical Code of Conduct of the Institute. e. Whenever I have used materials (data, theoretical analysis, figures and text) from other sources, I have given due credit to them by citing them in the text of the thesis and giving their details in the references. Further, I have taken permission from the copyright owners of the sources, whenever necessary. IJCRT2012039 International Journal of Creative Research Thoughts (IJCRT) www.ijcrt.org 284 www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 f. Whenever I have quoted written materials from other sources I have put them under quotation marks and given due credit to the sources by citing them and giving required details in the references. (Subhasis Basu) ACKNOWLEDGEMENT This preface is to extend an appreciation to all those individuals who with their generous co- operation guided us in every aspect to make this design and drawing successful. -

Histamine and Antihistamines Sites of Action Conditions Which Cause Release Aron H
Learning Objectives I Histamine Pharmacological effects Histamine and Antihistamines Sites of action Conditions which cause release Aron H. Lichtman, Ph.D. Diagnostic uses Associate Professor II Antihistamines acting at the H1 and H2 receptor Pharmacology and Toxicology Pharmacological effects Mechanisms of action Therapeutic uses Side effects and drug interactions Be familiar with the existence of the H3 receptor III Be able to describe the main mechanism of action of cromolyn sodium and its clinical uses Histamine Pharmacology First autacoid to be discovered. (Greek: autos=self; Histamine Formation akos=cure) Synthesized in 1907 Synthesized in mammalian tissues by Demonstrated to be a natural constituent of decarboxylation of the amino acid l-histidine mammalian tissues (1927) Involved in inflammatory and anaphylactic reactions. Local application causes swelling redness, and edema, mimicking a mild inflammatory reaction. Large systemic doses leads to profound vascular changes similar to those seen after shock or anaphylactic origin Histamine Stored in complex with: Heparin Chondroitin Sulfate Eosinophilic Chemotactic Factor Neutrophilic Chemotactic Factor Proteases 1 Conditions That Release Histamine 1. Tissue injury: Any physical or chemical agent that injures tissue, skin or mucosa are particularly sensitive to injury and will cause the immediate release of histamine from mast cells. 2. Allergic reactions: exposure of an antigen to a previously sensitized (exposed) subject can immediately trigger allergic reactions. If sensitized by IgE antibodies attached to their surface membranes will degranulate when exposed to the appropriate antigen and release histamine, ATP and other mediators. 3. Drugs and other foreign compounds: morphine, dextran, antimalarial drugs, dyes, antibiotic bases, alkaloids, amides, quaternary ammonium compounds, enzymes (phospholipase C). -

Mediated Multidrug Resistance (MDR) in Cancer
A Thesis Entitled: Differential Effects of Dopamine D3 Receptor Antagonists in Modulating ABCG2 - Mediated Multidrug Resistance (MDR) in Cancer by Noor A. Hussein Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Master of Science in Pharmaceutical Sciences Degree in Pharmacology and Toxicology _________________________________________ Dr. Amit K. Tiwari, Committee Chair _________________________________________ Dr. Frank Hall , Committee Member _________________________________________ Dr. Zahoor Shah, Committee Member _________________________________________ Dr. Caren Steinmiller, Committee Member _________________________________________ Dr. Amanda Bryant-Friedrich, Dean College of Graduate Studies The University of Toledo May 2017 Copyright 2017, Noor A. Hussein This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract of Differential Effects of Dopamine D3 Receptor Antagonists in Modulating ABCG2 - Mediated Multidrug Resistance (MDR) by Noor A. Hussein Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Master of Science in Pharmaceutical Sciences Degree in Pharmacology and Toxicology The University of Toledo May 2017 The G2 subfamily of the ATP-binding cassette transporters (ABCG2), also known as the breast cancer resistance protein (BRCP), is an efflux transporter that plays an important role in protecting the cells against endogenous and exogenous toxic substances. The ABCG2 transporters are also highly expressed in the blood brain barrier (BBB), providing protection against specific toxic compounds. Unfortunately, their overexpression in cancer cells results in the development of multi-drug resistance (MDR), and thus, chemotherapy failure. Dopamine3 receptor (D3R) antagonists were shown to have excellent anti-addiction properties in preclinical animal models but produced limited clinical success with the lead molecule. -
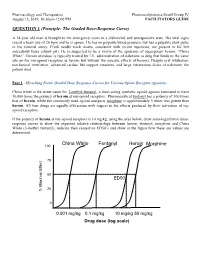
QUESTION 1 (Principle: the Graded Dose-Response Curve)
Pharmacology and Therapeutics Pharmacodynamics Small Group IV August 15, 2019, 10:30am-12:00 PM FACILITATORS GUIDE QUESTION 1 (Principle: The Graded Dose-Response Curve) A 34-year old man is brought to the emergency room in a disheveled and unresponsive state. His vital signs reveal a heart rate of 26 bpm and he is apneic. He has no palpable blood pressure, but has a palpable slow pulse in his femoral artery. Fresh needle track marks, consistent with recent injections, are present in his left antecubital fossa (elbow pit). He is suspected to be a victim of the epidemic of superpotent heroin, “China White”. Heroin overdose is typically treated by I.V. administration of naloxone (a drug that binds to the same site on the mu-opioid receptors as heroin, but without the narcotic effects of heroin). Despite oral intubation, mechanical ventilation, advanced cardiac life support measures, and large intravenous doses of naloxone, the patient died. Part 1 (Teaching Point: Graded Dose Response Curves for Various Opiate Receptor Agonists) China white is the street name for 3-methyl-fentanyl, a short-acting synthetic opioid agonist estimated to have 10,000 times the potency of heroin at mu-opioid receptors. Pharmaceutical fentanyl has a potency of 100 times that of heroin, while the commonly used opioid analgesic morphine is approximately 5 times less potent than heroin. All four drugs are equally efficacious with respect to the effects produced by their activation of m opioid receptors. If the potency of heroin at mu-opioid receptors is 10 mg/kg, using the axes below, draw semi-logarithmic dose- response curves to show the expected relative relationships between heroin, fentanyl, morphine and China White (3-methyl fentanyl), indicate their respective ED50’s and show in the figure how these are values are determined.