Role of Negative Regulation of Immune Signaling Pathways in Neutrophil Function
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Gene Expression Within the Periaqueductal Gray Is Linked to Vocal Behavior and Early- Onset Parkinsonism in Pink1 Knockout Rats Cynthia A
Kelm-Nelson and Gammie BMC Genomics (2020) 21:625 https://doi.org/10.1186/s12864-020-07037-4 RESEARCH ARTICLE Open Access Gene expression within the periaqueductal gray is linked to vocal behavior and early- onset parkinsonism in Pink1 knockout rats Cynthia A. Kelm-Nelson1* and Stephen Gammie2 Abstract Background: Parkinson’s disease (PD) is a degenerative disease with early-stage pathology hypothesized to manifest in brainstem regions. Vocal deficits, including soft, monotone speech, result in significant clinical and quality of life issues and are present in 90% of PD patients; yet the underlying pathology mediating these significant voice deficits is unknown. The Pink1−/− rat is a valid model of early-onset PD that presents with analogous vocal communication deficits. Previous work shows abnormal α-synuclein protein aggregation in the periaqueductal gray (PAG), a brain region critical and necessary to the modulation of mammalian vocal behavior. In this study, we used high-throughput RNA sequencing to examine gene expression within the PAG of both male and female Pink1−/− rats as compared to age-matched wildtype controls. We used a bioinformatic approach to (1) test the hypothesis that loss of Pink1 in the PAG will influence the differential expression of genes that interact with Pink1, (2) highlight other key genes that relate to this type of Mendelian PD, and (3) catalog molecular targets that may be important for the production of rat vocalizations. Results: Knockout of the Pink1 gene resulted in differentially expressed genes for both male and female rats that also mapped to human PD datasets. Pathway analysis highlighted several significant metabolic pathways. -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -

Technical Note, Appendix: an Analysis of Blood Processing Methods to Prepare Samples for Genechip® Expression Profiling (Pdf, 1
Appendix 1: Signature genes for different blood cell types. Blood Cell Type Source Probe Set Description Symbol Blood Cell Type Source Probe Set Description Symbol Fraction ID Fraction ID Mono- Lympho- GSK 203547_at CD4 antigen (p55) CD4 Whitney et al. 209813_x_at T cell receptor TRG nuclear cytes gamma locus cells Whitney et al. 209995_s_at T-cell leukemia/ TCL1A Whitney et al. 203104_at colony stimulating CSF1R lymphoma 1A factor 1 receptor, Whitney et al. 210164_at granzyme B GZMB formerly McDonough (granzyme 2, feline sarcoma viral cytotoxic T-lymphocyte- (v-fms) oncogene associated serine homolog esterase 1) Whitney et al. 203290_at major histocompatibility HLA-DQA1 Whitney et al. 210321_at similar to granzyme B CTLA1 complex, class II, (granzyme 2, cytotoxic DQ alpha 1 T-lymphocyte-associated Whitney et al. 203413_at NEL-like 2 (chicken) NELL2 serine esterase 1) Whitney et al. 203828_s_at natural killer cell NK4 (H. sapiens) transcript 4 Whitney et al. 212827_at immunoglobulin heavy IGHM Whitney et al. 203932_at major histocompatibility HLA-DMB constant mu complex, class II, Whitney et al. 212998_x_at major histocompatibility HLA-DQB1 DM beta complex, class II, Whitney et al. 204655_at chemokine (C-C motif) CCL5 DQ beta 1 ligand 5 Whitney et al. 212999_x_at major histocompatibility HLA-DQB Whitney et al. 204661_at CDW52 antigen CDW52 complex, class II, (CAMPATH-1 antigen) DQ beta 1 Whitney et al. 205049_s_at CD79A antigen CD79A Whitney et al. 213193_x_at T cell receptor beta locus TRB (immunoglobulin- Whitney et al. 213425_at Homo sapiens cDNA associated alpha) FLJ11441 fis, clone Whitney et al. 205291_at interleukin 2 receptor, IL2RB HEMBA1001323, beta mRNA sequence Whitney et al. -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

CBA Vs. BL/6 BL/6 Vs. CBA Biological Processes
Table SI. Gene ontology (GO) analysis of genes preferentially expressed by CBA or BL/6 DCs CBA vs. BL/6 BL/6 vs. CBA Biological • Immune Response • Regulation of immune effector Processes • Defense response process o Innate immune response o Regulation of leukocyte • Response to external stimulus mediated immunity . Regulation of lymphocyte mediated immunity • Amino sugar catabolic process o Glucosamine catabolic process . N-acetylglucosamine catabolic process • Chitin metabolic process o Chitin catabolic process • Defense response • Response to wounding o Inflammatory response Cellular • Intrinsic to membrane • Extracellular space Component • Cell surface • External side of plasma membrane Molecular • Molecular transducer activity • Chitinase activity Function o Signal transducer activity • G-protein-coupled receptor binding . Receptor activity o Chemokine receptor binding • Carbohydrate binding • Cytokine activity o Sugar binding o Chemokine activity . Monosaccharide binding • C-C chemokine binding • C-C chemokine receptor activity • Cytokine binding 1 Table SII. Genes preferentially expressed by unstimulated CBA DCs mRNA Access Fold Gene Symbol Number Diff mRNA Description H2-Ea NM 010381 88.23 Histocompatibility 2, class II antigen E alpha Ifi202b NM 008327 32.73 Interferon activated gene 202B Chi3l1 NM 007695 24.14 Chitinase-3-like 1 Ctse NM 007799 23.64 Cathepsin E Cd209a (SIGNR5) NM 133238 18.32 CD209a antigen Colec12 (CL-P1) NM130449 16.95 Collectin sub-family member 12 Retnla (FIZZ1) NM 020509 10.22 resistin like alpha Cd209c -

Integrated Weighted Gene Co-Expression Network Analysis Identi Ed That TLR2 and CD40 Are Related to Coronary Artery Disease
Integrated weighted gene co-expression network analysis identied that TLR2 and CD40 are related to coronary artery disease Bin Qi Liuzhou People's Hospital Jian-Hong Chen Liuzhou People's Hospital Lin Tao Liuzhou People's Hospital Chuan-Meng Zhu Liuzhou People's Hospital Yong Wang Liuzhou People's Hospital Guo-Xiong Deng The First People's of Nanning Liu Miao ( [email protected] ) LiuZhou People's Hospital https://orcid.org/0000-0001-6642-7005 Research article Keywords: Gene Expression Omnibus, Integrated weighted gene co-expression network analysis (WGCNA), Functional enrichment, Functional validation and prognostic analysis. Posted Date: October 7th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-86115/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/16 Abstract Background: The current research attempted to identify possible hub genes and pathways of coronary artery disease (CAD) and to detect the possible mechanisms. Methods: Array data from GSE90074 were downloaded from the Gene Expression Omnibus (GEO) database. Integrated weighted gene co-expression network analysis (WGCNA) was performed to analyze the gene module and clinical characteristics. Gene Ontology annotation, Disease Ontology and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed by clusterProler and the DOSE package in R. A protein-protein interaction (PPI) network was established using Cytoscape software, and signicant modules were analyzed using Molecular Complex Detection to identify hub genes. Then, further functional validation of hub genes in other microarrays and population samples was performed, and survival analysis was performed to investigate the prognosis. -

Ncounter Fibrosis Panel
PRODUCT BULLETIN nCounter® Fibrosis Panel Gene Expression Panel Four Stages of Fibrosis • Disease Pathogenesis • Biomarkers of Progression Uncover the mechanisms of disease pathogenesis, identify biomarkers of progression, and develop signatures for therapeutic response with the nCounter Fibrosis Panel. This gene expression panel combines hundreds of genes involved in the initial tissue damage response, chronic inflammation, proliferation of pro-fibrotic cells, and tissue modification that leads to fibrotic disease of the lungs, heart, liver, kidney, and skin. INITIATION Product Highlights • Profile 770 genes across 51 annotated pathways involved in the four stages of fibrosis T is s s • Initiation s u e e r I • t D N Inflammation N S a F l m • Proliferation O l L I e a A g • Modification T C M e A M C • I Study pathogenesis and identify biomarkers for fibrotic A F I T diseases of the lungs, heart, liver, kidney, and skin I D O O N • Understand the signaling cascade from cell M stress to inflammation • Quantify the relative abundance of 14 different W g ound Healin immune cell types • Identify biomarkers of therapeutic response PR N OLIFERATIO Feature Specifications Number of Targets 770 (Human), 770 (Mouse), including internal reference genes Standard Input Material (No amplification required) 25-300 ng Sample Input - Low Input As little as 1 ng with nCounter Low Input Kit (sold separately) Cultured cells/cell lysates, sorted cells, FFPE-derived RNA, total RNA, Sample Type(s) fragmented RNA, PBMCs, and whole blood/plasma Add up to 55 unique genes with Panel-Plus and up to 10 custom Customizable protein targets Time to Results Approximately 24 hours Data Analysis nSolver™ Analysis Software (RUO) FOR RESEARCH USE ONLY. -

Functionalization of Brain Region-Specific Spheroids With
www.nature.com/scientificreports OPEN Functionalization of Brain Region- specifc Spheroids with Isogenic Microglia-like Cells Received: 5 February 2019 Liqing Song1,6, Xuegang Yuan1, Zachary Jones2, Cynthia Vied 3, Yu Miao1, Mark Marzano1, Accepted: 15 July 2019 Thien Hua4, Qing-Xiang Amy Sang4,5, Jingjiao Guan1, Teng Ma1, Yi Zhou2 & Yan Li1,5 Published: xx xx xxxx Current brain spheroids or organoids derived from human induced pluripotent stem cells (hiPSCs) still lack a microglia component, the resident immune cells in the brain. The objective of this study is to engineer brain region-specifc organoids from hiPSCs incorporated with isogenic microglia- like cells in order to enhance immune function. In this study, microglia-like cells were derived from hiPSCs using a simplifed protocol with stage-wise growth factor induction, which expressed several phenotypic markers, including CD11b, IBA-1, CX3CR1, and P2RY12, and phagocytosed micron-size super-paramagnetic iron oxides. The derived cells were able to upregulate pro-infammatory gene (TNF-α) and secrete anti-infammatory cytokines (i.e., VEGF, TGF-β1, and PGE2) when stimulated with amyloid β42 oligomers, lipopolysaccharides, or dexamethasone. The derived isogenic dorsal cortical (higher expression of TBR1 and PAX6) and ventral (higher expression of NKX2.1 and PROX1) spheroids/ organoids displayed action potentials and synaptic activities. Co-culturing the microglia-like cells (MG) with the dorsal (D) or ventral (V) organoids showed diferential migration ability, intracellular Ca2+ signaling, and the response to pro-infammatory stimuli (V-MG group had higher TNF-α and TREM2 expression). Transcriptome analysis exhibited 37 microglia-related genes that were diferentially expressed in MG and D-MG groups. -

Myeloid Innate Immunity Mouse Vapril2018
Official Symbol Accession Alias / Previous Symbol Official Full Name 2810417H13Rik NM_026515.2 p15(PAF), Pclaf RIKEN cDNA 2810417H13 gene 2900026A02Rik NM_172884.3 Gm449, LOC231620 RIKEN cDNA 2900026A02 gene Abcc8 NM_011510.3 SUR1, Sur, D930031B21Rik ATP-binding cassette, sub-family C (CFTR/MRP), member 8 Acad10 NM_028037.4 2410021P16Rik acyl-Coenzyme A dehydrogenase family, member 10 Acly NM_134037.2 A730098H14Rik ATP citrate lyase Acod1 NM_008392.1 Irg1 aconitate decarboxylase 1 Acot11 NM_025590.4 Thea, 2010309H15Rik, 1110020M10Rik,acyl-CoA Them1, thioesterase BFIT1 11 Acot3 NM_134246.3 PTE-Ia, Pte2a acyl-CoA thioesterase 3 Acox1 NM_015729.2 Acyl-CoA oxidase, AOX, D130055E20Rikacyl-Coenzyme A oxidase 1, palmitoyl Adam19 NM_009616.4 Mltnb a disintegrin and metallopeptidase domain 19 (meltrin beta) Adam8 NM_007403.2 CD156a, MS2, E430039A18Rik, CD156a disintegrin and metallopeptidase domain 8 Adamts1 NM_009621.4 ADAM-TS1, ADAMTS-1, METH-1, METH1a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 1 Adamts12 NM_175501.2 a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 12 Adamts14 NM_001081127.1 Adamts-14, TS14 a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 14 Adamts17 NM_001033877.4 AU023434 a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 17 Adamts2 NM_001277305.1 hPCPNI, ADAM-TS2, a disintegrin and ametalloproteinase disintegrin-like and with metallopeptidase thrombospondin -
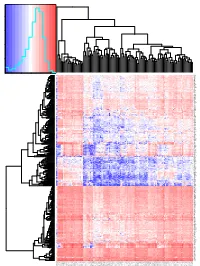
R Graphics Output
Color Key and Histogram 8000 6000 Count 4000 2000 0 0.4 0.6 0.8 1 1.2 Value NLRC4 CXorf21 SNX20 KMO LILRA5 CFP TIGIT FOXP3 CTLA4 KLRK1 SLA2 KLRB1 AFF3 P2RX5 SLAMF1 SIT1 LY9 EBI3 SIRPG KIAA0748 GPR171 GRAP2 SCML4 CD8B CHI3L2 ADAMDEC1 TNFSF8 LAX1 CD38 CD28 SH2D1A NCF1C C17orf87 NCF1B FCRL5 NELL2 CCR2 P2RY13 TLR7 TLR8 SIGLEC7 LILRA2 SIGLEC5 GAPT RGS18 FAM70A SIGLEC8 ART4 CLEC4E GZMH CD180 LILRB1 NCF1 IGSF6 TFEC CD300LF TNFAIP8L2 DPEP2 GPR65 CLEC4A NLRP3 CCL4 CCL3 IL21R AQP9 LILRA6 CD209 LILRB5 FCN1 C19orf59 CCL13 SIGLEC14 C16orf89 CADM3 FABP4 RELN PTX3 SCN7A PLP1 CD1C CD1E CCL17 PI16 SCARA5 SELE NRXN1 RAB39B GPM6A PRPH SLC16A12 P2RY10 PYHIN1 CD3G THEMIS KCNA3 ZNF831 TBX21 CD40LG UBASH3A GZMM TLR10 PNOC C8orf80 CXCR5 PLA2G2D FCRL3 BTLA TRAT1 EOMES IKZF3 P2RY12 ACSM5 EMR1 CEACAM4 CTSG FLT3 GHRL MADCAM1 LCN10 MAL PLIN1 CASS4 P2RY14 CYSLTR1 C10orf128 CD226 ICOS CCR4 FPR2 CXCR2 MEFV CEACAM21 CRTAM DNAJC5B PDE6G CLEC12A FCGR3B CCL8 HAS1 MS4A1 BLK CR2 CXCL13 C4orf7 RFX6 SVOP TMEM132D SH3GL2 SLC7A14 TAGLN3 KCNH6 RXRG SSTR3 KCNMB2 UNC80 FLJ43390 SLC4A10 SEZ6L CALY ANGPTL7 NEUROD1 CELF3 SYT4 GAD2 G6PC2 IAPP SERPINB3 TNNT1 SPRR1A PNCK ABCD2 GPR174 TARP LILRA1 GPR141 CCR8 DTHD1 CXCR2P1 TIFAB ANKRD55 CD1B TIMD4 LILRA3 HLA−DPB2 CLECL1 LILRA4 KEL CCL23 EMR3 DNASE1L3 VIT XPNPEP2 CNTN2 CLEC9A LGALS12 LTA NCR3 GPR18 STAP1 GP1BA C5orf20 TNFRSF17 FCRLA KIAA0125 CD19 FCRL2 VPREB3 FAM129C TNFRSF13B PAX5 FCRL1 CNR2 TCL1A FCER2 CLEC17A BEND4 KLHL14 PKHD1L1 C13orf36 CDH10 CALB1 NPHS1 TCL6 LBP LEP ADIPOQ TUSC5 CD300LG CIDEA KIAA0408 SASH3 ITGAL PLEK RCSD1 IKZF1 GIMAP5