Case Reports Ultrasound Relevance in Prenatal Diagnosis of Vacterl
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

VATER/VACTERL Association in Palestinian Children: a Case Report
www.symbiosisonline.org Symbiosis www.symbiosisonlinepublishing.com Research Article International Journal of Pediatrics & Child Care Open Access VATER/VACTERL Association in Palestinian Children: A Case Report Basal A Ahmed1, Elessi Khamis2* 1Specialist and Head of Pediatrics, Shaheed Mohammed Al - Durra Hospital 2Assistant Professor, Faculty of medicine, Islamic university- Gaza Received:December 13, 2017; Accepted: February 3, 2018; Published: February 6, 2018 *Corresponding author: Elessi Khamis, MD Pediatrics, Assistant Professor, Faculty of medicine, Islamic university- Gaza, E-mail: khamis_essi@yahoo. com have reported a prevalence among infants of one in 10 000 to one Abstract of at least three of the following congenital malformations: vertebral however,in 40 000 live-bornchromosomal infants abnormalities (approximately have <1-9/100,000 also been described infants) VACTERL/VATER association is typically defined by the presence [2]. Most of the cases of VACTERL association occur sporadically; defects, anal atresia, cardiac defects, tracheo-esophageal fistula, stress and usage of oral contraceptives at the initial stages of by evidence linking all of the human disease genes for the VATER/ in a few cases [3]. Maternal diabetes, teratogenic drugs, physical renal anomalies, and limb abnormalities. This finding is supported pregnancy have been suggested as possible causes [4]. VACTERL is believed to result from an early embryonic insult, more VACTERL association identified to date, namely, FGF8, FOXF1, HOXD13, LPP, TRAP1, and ZIC3, with renal malformations. VATER association was first described in 1972 by Quan and Smith. We present here a specifically of blastogenic origin occurring during the first 4 75 days male boy with cardiac (VSD, PDA), esophageal atresia, anal weeks of embryogenesis, so the expected effects are primary, abnormalities (sacral dimple), and genitourinary (hypospadias and polytopic,This early developmental embryonic field event defects can [5]lead (Figure to different 1). -

Holt-Oram Syndrome: a Clinical Genetic Study J Med Genet: First Published As 10.1136/Jmg.33.4.300 on 1 April 1996
300_0fMed Genet 1996;33:300-307 Holt-Oram syndrome: a clinical genetic study J Med Genet: first published as 10.1136/jmg.33.4.300 on 1 April 1996. Downloaded from R A Newbury-Ecob, R Leanage, J A Raebum, I D Young Abstract to clarify the spectrum of abnormalities and to A clinical and genetic study of the Holt- delineate the HOS phenotype led us to review Oram syndrome (HOS) has been carried the clinical features in our patients, and dis- out in the United Kingdom involving 55 tinguish the clinical features most helpful for cases designated Holt-Oram syndrome, counselling purposes. together with their parents and sibs. Data This study was carried out in conjunction from the clinical assessment of both fa- with a genetic linkage study which has shown milial and isolated cases were used to de- genetic heterogeneity in the Holt-Oram syn- fine the HOS phenotype and to outline drome, with one gene (HOS1) being localised the spectrum of abnormalities, especially to chromosome 12 in five out ofseven families.7 factors affecting severity. Skeletal defects No phenotypic differences could be detected affected the upper limbs exclusively and between the linked and unlinked families. were bilateral and asymmetrical. They ranged from minor signs such as clino- dactyly, limited supination, and sloping Patients and methods shoulders to severe reduction deformities The study was carried out between March 1991 of the upper arm (4.5%). The radial ray and September 1993. Cases were ascertained was predominantly affected and the left by contacting clinical geneticists and paediatric side was more severely affected than the cardiologists and through the support group right. -

Case Report Upper Limb Meromelia with Oligodactyly and Brachymesophalangy of the Foot: an Unusual Association
Hindawi Case Reports in Radiology Volume 2019, Article ID 3419383, 5 pages https://doi.org/10.1155/2019/3419383 Case Report Upper Limb Meromelia with Oligodactyly and Brachymesophalangy of the Foot: An Unusual Association Meltem Özdemir , Rasime Pelin Kavak , and Önder Eraslan University of Health Sciences, Dıs¸kapı Yıldırım Beyazıt Training and Research Hospital, Department of Radiology, Ankara, Turkey Correspondence should be addressed to Meltem Ozdemir;¨ [email protected] Received 1 May 2019; Accepted 7 June 2019; Published 24 June 2019 Academic Editor: Ravi Bhargava Copyright © 2019 Meltem Ozdemir¨ et al. Tis is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Meromelia is a rare skeletal abnormality characterized by the partial absence of at least one limb. Several mechanisms have been postulated to explain the etiopathogenesis of the disorder. Most of the cases of meromelia are reported to be sporadic. It can occur either in isolation or with other congenital malformations. VACTERL association, gastroschisis, atrial septal defect, proximal femoral focal defciency, and fbular hemimelia are the congenital abnormalities reported to be in association with meromelia. However, no other congenital abnormalities in association with meromelia have been recorded to date. We herein present an unusual case of bilateral upper limb meromelia accompanied by unilateral oligodactyly and brachymesophalangy of the foot. 1. Introduction herein present an unusual case of meromelia accompanied by congenital deformity of the foot. Amelia refers to the complete absence of at least one limb, and meromelia is characterized by the partial absence of at least one limb. -

VACTERL/VATER Association Benjamin D Solomon
Solomon Orphanet Journal of Rare Diseases 2011, 6:56 http://www.ojrd.com/content/6/1/56 REVIEW Open Access VACTERL/VATER Association Benjamin D Solomon Abstract VACTERL/VATER association is typically defined by the presence of at least three of the following congenital malformations: vertebral defects, anal atresia, cardiac defects, tracheo-esophageal fistula, renal anomalies, and limb abnormalities. In addition to these core component features, patients may also have other congenital anomalies. Although diagnostic criteria vary, the incidence is estimated at approximately 1 in 10,000 to 1 in 40,000 live-born infants. The condition is ascertained clinically by the presence of the above-mentioned malformations; importantly, there should be no clinical or laboratory-based evidence for the presence of one of the many similar conditions, as the differential diagnosis is relatively large. This differential diagnosis includes (but is not limited to) Baller-Gerold syndrome, CHARGE syndrome, Currarino syndrome, deletion 22q11.2 syndrome, Fanconi anemia, Feingold syndrome, Fryns syndrome, MURCS association, oculo-auriculo-vertebral syndrome, Opitz G/BBB syndrome, Pallister- Hall syndrome, Townes-Brocks syndrome, and VACTERL with hydrocephalus. Though there are hints regarding causation, the aetiology has been identified only in a small fraction of patients to date, likely due to factors such as a high degree of clinical and causal heterogeneity, the largely sporadic nature of the disorder, and the presence of many similar conditions. New genetic research methods offer promise that the causes of VACTERL association will be better defined in the relatively near future. Antenatal diagnosis can be challenging, as certain component features can be difficult to ascertain prior to birth. -

Early ACCESS Diagnosed Conditions List
Iowa Early ACCESS Diagnosed Conditions Eligibility List List adapted with permission from Early Intervention Colorado To search for a specific word type "Ctrl F" to use the "Find" function. Is this diagnosis automatically eligible for Early Medical Diagnosis Name Other Names for the Diagnosis and Additional Diagnosis Information ACCESS? 6q terminal deletion syndrome Yes Achondrogenesis I Parenti-Fraccaro Yes Achondrogenesis II Langer-Saldino Yes Schinzel Acrocallosal syndrome; ACLS; ACS; Hallux duplication, postaxial polydactyly, and absence of the corpus Acrocallosal syndrome, Schinzel Type callosum Yes Acrodysplasia; Arkless-Graham syndrome; Maroteaux-Malamut syndrome; Nasal hypoplasia-peripheral dysostosis-intellectual disability syndrome; Peripheral dysostosis-nasal hypoplasia-intellectual disability (PNM) Acrodysostosis syndrome Yes ALD; AMN; X-ALD; Addison disease and cerebral sclerosis; Adrenomyeloneuropathy; Siemerling-creutzfeldt disease; Bronze schilder disease; Schilder disease; Melanodermic Leukodystrophy; sudanophilic leukodystrophy; Adrenoleukodystrophy Pelizaeus-Merzbacher disease Yes Agenesis of Corpus Callosum Absence of the corpus callosum; Hypogenesis of the corpus callosum; Dysplastic corpus callosum Yes Agenesis of Corpus Callosum and Chorioretinal Abnormality; Agenesis of Corpus Callosum With Chorioretinitis Abnormality; Agenesis of Corpus Callosum With Infantile Spasms And Ocular Anomalies; Chorioretinal Anomalies Aicardi syndrome with Agenesis Yes Alexander Disease Yes Allan Herndon syndrome Allan-Herndon-Dudley -

Whole-Exome Sequencing Identifies Causative Mutations in Families
BASIC RESEARCH www.jasn.org Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract Amelie T. van der Ven,1 Dervla M. Connaughton,1 Hadas Ityel,1 Nina Mann,1 Makiko Nakayama,1 Jing Chen,1 Asaf Vivante,1 Daw-yang Hwang,1 Julian Schulz,1 Daniela A. Braun,1 Johanna Magdalena Schmidt,1 David Schapiro,1 Ronen Schneider,1 Jillian K. Warejko,1 Ankana Daga,1 Amar J. Majmundar,1 Weizhen Tan,1 Tilman Jobst-Schwan,1 Tobias Hermle,1 Eugen Widmeier,1 Shazia Ashraf,1 Ali Amar,1 Charlotte A. Hoogstraaten,1 Hannah Hugo,1 Thomas M. Kitzler,1 Franziska Kause,1 Caroline M. Kolvenbach,1 Rufeng Dai,1 Leslie Spaneas,1 Kassaundra Amann,1 Deborah R. Stein,1 Michelle A. Baum,1 Michael J.G. Somers,1 Nancy M. Rodig,1 Michael A. Ferguson,1 Avram Z. Traum,1 Ghaleb H. Daouk,1 Radovan Bogdanovic,2 Natasa Stajic,2 Neveen A. Soliman,3,4 Jameela A. Kari,5,6 Sherif El Desoky,5,6 Hanan M. Fathy,7 Danko Milosevic,8 Muna Al-Saffar,1,9 Hazem S. Awad,10 Loai A. Eid,10 Aravind Selvin,11 Prabha Senguttuvan,12 Simone Sanna-Cherchi,13 Heidi L. Rehm,14 Daniel G. MacArthur,14,15 Monkol Lek,14,15 Kristen M. Laricchia,15 Michael W. Wilson,15 Shrikant M. Mane,16 Richard P. Lifton,16,17 Richard S. Lee,18 Stuart B. Bauer,18 Weining Lu,19 Heiko M. Reutter ,20,21 Velibor Tasic,22 Shirlee Shril,1 and Friedhelm Hildebrandt1 Due to the number of contributing authors, the affiliations are listed at the end of this article. -

Appendix 3.1 Birth Defects Descriptions for NBDPN Core, Recommended, and Extended Conditions Updated March 2017
Appendix 3.1 Birth Defects Descriptions for NBDPN Core, Recommended, and Extended Conditions Updated March 2017 Participating members of the Birth Defects Definitions Group: Lorenzo Botto (UT) John Carey (UT) Cynthia Cassell (CDC) Tiffany Colarusso (CDC) Janet Cragan (CDC) Marcia Feldkamp (UT) Jamie Frias (CDC) Angela Lin (MA) Cara Mai (CDC) Richard Olney (CDC) Carol Stanton (CO) Csaba Siffel (GA) Table of Contents LIST OF BIRTH DEFECTS ................................................................................................................................................. I DETAILED DESCRIPTIONS OF BIRTH DEFECTS ...................................................................................................... 1 FORMAT FOR BIRTH DEFECT DESCRIPTIONS ................................................................................................................................. 1 CENTRAL NERVOUS SYSTEM ....................................................................................................................................... 2 ANENCEPHALY ........................................................................................................................................................................ 2 ENCEPHALOCELE ..................................................................................................................................................................... 3 HOLOPROSENCEPHALY............................................................................................................................................................. -

Genetics of Atrioventricular Canal Defects Flaminia Pugnaloni1, Maria Cristina Digilio2, Carolina Putotto1, Enrica De Luca1, Bruno Marino1 and Paolo Versacci1*
Pugnaloni et al. Italian Journal of Pediatrics (2020) 46:61 https://doi.org/10.1186/s13052-020-00825-4 REVIEW Open Access Genetics of atrioventricular canal defects Flaminia Pugnaloni1, Maria Cristina Digilio2, Carolina Putotto1, Enrica De Luca1, Bruno Marino1 and Paolo Versacci1* Abstract Atrioventricular canal defect (AVCD) represents a quite common congenital heart defect (CHD) accounting for 7.4% of all cardiac malformations. AVCD is a very heterogeneous malformation that can occur as a phenotypical cardiac aspect in the context of different genetic syndromes but also as an isolated, non-syndromic cardiac defect. AVCD has also been described in several pedigrees suggesting a pattern of familiar recurrence. Targeted Next Generation Sequencing (NGS) techniques are proved to be a powerful tool to establish the molecular heterogeneity of AVCD. Given the complexity of cardiac embryology, it is not surprising that multiple genes deeply implicated in cardiogenesis have been described mutated in patients with AVCD. This review attempts to examine the recent advances in understanding the molecular basis of this complex CHD in the setting of genetic syndromes or in non- syndromic patients. Keywords: Congenital heart disease, Atrioventricular canal defect, Genetics Introduction extracellular matrix, leading to absent or incomplete fu- The atrioventricular canal defect (AVCD), also called sion of ventral (antero-superior) and dorsal (postero-in- atrioventricular septal defect, is a quite common con- ferior) atrioventricular cushions [2–4]. Nevertheless, the genital heart defect (CHD), accounting for 7.4% of all hypothesis that extracardiac progenitor cells contribute cardiac malformations. It can be anatomically classified also to the growth of the inlet part of the heart has been in complete, partial and intermediate types. -

Platform Abstracts
American Society of Human Genetics 65th Annual Meeting October 6–10, 2015 Baltimore, MD PLATFORM ABSTRACTS Wednesday, October 7, 9:50-10:30am Abstract #’s Friday, October 9, 2:15-4:15 pm: Concurrent Platform Session D: 4. Featured Plenary Abstract Session I Hall F #1-#2 46. Hen’s Teeth? Rare Variants and Common Disease Ballroom I #195-#202 Wednesday, October 7, 2:30-4:30pm Concurrent Platform Session A: 47. The Zen of Gene and Variant 15. Update on Breast and Prostate Assessment Ballroom III #203-#210 Cancer Genetics Ballroom I #3-#10 48. New Genes and Mechanisms in 16. Switching on to Regulatory Variation Ballroom III #11-#18 Developmental Disorders and 17. Shedding Light into the Dark: From Intellectual Disabilities Room 307 #211-#218 Lung Disease to Autoimmune Disease Room 307 #19-#26 49. Statistical Genetics: Networks, 18. Addressing the Difficult Regions of Pathways, and Expression Room 309 #219-#226 the Genome Room 309 #27-#34 50. Going Platinum: Building a Better 19. Statistical Genetics: Complex Genome Room 316 #227-#234 Phenotypes, Complex Solutions Room 316 #35-#42 51. Cancer Genetic Mechanisms Room 318/321 #235-#242 20. Think Globally, Act Locally: Copy 52. Target Practice: Therapy for Genetic Hilton Hotel Number Variation Room 318/321 #43-#50 Diseases Ballroom 1 #243-#250 21. Recent Advances in the Genetic Basis 53. The Real World: Translating Hilton Hotel of Neuromuscular and Other Hilton Hotel Sequencing into the Clinic Ballroom 4 #251-#258 Neurodegenerative Phenotypes Ballroom 1 #51-#58 22. Neuropsychiatric Diseases of Hilton Hotel Friday, October 9, 4:30-6:30pm Concurrent Platform Session E: Childhood Ballroom 4 #59-#66 54. -

EUROCAT Syndrome Guide
JRC - Central Registry european surveillance of congenital anomalies EUROCAT Syndrome Guide Definition and Coding of Syndromes Version July 2017 Revised in 2016 by Ingeborg Barisic, approved by the Coding & Classification Committee in 2017: Ester Garne, Diana Wellesley, David Tucker, Jorieke Bergman and Ingeborg Barisic Revised 2008 by Ingeborg Barisic, Helen Dolk and Ester Garne and discussed and approved by the Coding & Classification Committee 2008: Elisa Calzolari, Diana Wellesley, David Tucker, Ingeborg Barisic, Ester Garne The list of syndromes contained in the previous EUROCAT “Guide to the Coding of Eponyms and Syndromes” (Josephine Weatherall, 1979) was revised by Ingeborg Barisic, Helen Dolk, Ester Garne, Claude Stoll and Diana Wellesley at a meeting in London in November 2003. Approved by the members EUROCAT Coding & Classification Committee 2004: Ingeborg Barisic, Elisa Calzolari, Ester Garne, Annukka Ritvanen, Claude Stoll, Diana Wellesley 1 TABLE OF CONTENTS Introduction and Definitions 6 Coding Notes and Explanation of Guide 10 List of conditions to be coded in the syndrome field 13 List of conditions which should not be coded as syndromes 14 Syndromes – monogenic or unknown etiology Aarskog syndrome 18 Acrocephalopolysyndactyly (all types) 19 Alagille syndrome 20 Alport syndrome 21 Angelman syndrome 22 Aniridia-Wilms tumor syndrome, WAGR 23 Apert syndrome 24 Bardet-Biedl syndrome 25 Beckwith-Wiedemann syndrome (EMG syndrome) 26 Blepharophimosis-ptosis syndrome 28 Branchiootorenal syndrome (Melnick-Fraser syndrome) 29 CHARGE -

Blueprint Genetics Ciliopathy Panel
Ciliopathy Panel Test code: KI0701 Is a 107 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of Bardet-Biedl syndrome, Joubert syndrome, Meckel syndrome, nephronophthisis with or without retinal dystrophy, or complex ciliopathy phenotype. Isn’t ideal for a patient with primary ciliary dyskinesia or isomerism/heterotaxy. For patients with a suspicion of primary ciliary dyskinesia, Primary Ciliary Dyskinesia Panel is recommended. For patients with isomerism/heterotaxy, Heterotaxy and Situs Inversus Panel is recommended. About Ciliopathy Ciliopathies are a group of disorders resulting from either abnormal formation or function of cilia. Mutations in ciliary gene are known to cause single organ phenotypes, as well as complex syndromes. Ciliopathies have a broad range of phenotypes encompassing a number of different autosomal recessive, dominant and X-linked syndromes. As cilia are a component of almost all cells, ciliary dysfunction can manifest as a collection of features that include retinal degeneration, renal disease and brain malformations. Additional features may include congenital fibrocystic diseases of the liver and pancreas, diabetes, obesity and skeletal dysplasias. Ciliopathies can result from a mutation at a single locus in one patient while mutations affecting a number of different loci can, at the same time, can result in a similar phenotype in other patients. Ciliopathies can be classified according to whether there is aberrant function in an intact cilium or complete -
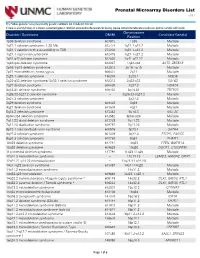
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome