Retinoic Acid Enhances the Expression of Interferon-Induced Proteins: Evidence for Multiple Mechanisms of Action
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Antibody-Based Delivery of Cytokine Payloads to Carbonic Anhydrase IX
Published OnlineFirst June 18, 2019; DOI: 10.1158/1535-7163.MCT-18-1301 Large Molecule Therapeutics Molecular Cancer Therapeutics Antibody-Based Delivery of Cytokine Payloads to Carbonic Anhydrase IX Leads to Cancer Cures in Immunocompetent Tumor-Bearing Mice Barbara Ziffels1, Marco Stringhini1, Philipp Probst1, Tim Fugmann2, Theo Sturm2, and Dario Neri1 Abstract Antibody–cytokine fusion proteins can have the potential TNF, IL2, or IL12 as payloads cured all mice in their therapy to increase the density and activity of subsets of leukocytes groups, whereas only a subset of mice was cured by the within the tumor mass. Here, we describe the design, produc- antibody-based delivery of IFNa2. Although the antibody tion, and characterization of four novel antibody–cytokine fusion with TNF mediated a rapid hemorrhagic necrosis of fusion proteins directed against human carbonic anhydrase IX, the tumor mass, a slower regression of the neoplastic lesions a highly validated marker of hypoxia that is overexpressed in (which continued after the last injection) was observed with clear cell renal cell carcinoma and other malignancies. As the other fusion proteins, and treated mice acquired protective immunomodulatory payloads we used TNF, IL2, IFNa2 (cor- anticancer immunity. A high proportion of tumor-infiltrating þ responding to products that are in clinical use), and IL12 (as CD8 T cells was specific to the retroviral antigen AH1; this cytokine potently activates T cells and NK cells). Therapy however, the LGPGREYRAL peptide derived from human experiments were performed in BALB/c mice, bearing CT26 carbonic anhydrase IX was also present on tumor cells. The tumors transfected with human carbonic anhydrase IX, in results described herein provide a rationale for the clinical use order to assess the performance of the fusion proteins in an of fully human antibody–cytokine fusions specific to carbonic immunocompetent setting. -

Mechanism of Action Through an IFN Type I-Independent Responses To
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 is online at: average * The Journal of Immunology , 12 of which you can access for free at: 2012; 188:3088-3098; Prepublished online 20 from submission to initial decision 4 weeks from acceptance to publication February 2012; doi: 10.4049/jimmunol.1101764 http://www.jimmunol.org/content/188/7/3088 MF59 and Pam3CSK4 Boost Adaptive Responses to Influenza Subunit Vaccine through an IFN Type I-Independent Mechanism of Action Elena Caproni, Elaine Tritto, Mario Cortese, Alessandro Muzzi, Flaviana Mosca, Elisabetta Monaci, Barbara Baudner, Anja Seubert and Ennio De Gregorio J Immunol cites 33 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2012/02/21/jimmunol.110176 4.DC1 This article http://www.jimmunol.org/content/188/7/3088.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 -

Brain Sciences
brain sciences Article Differential Expression of Genes Related to Innate Immune Responses in Ex Vivo Spinal Cord and Cerebellar Slice Cultures Infected with West Nile Virus Parminder J. S. Vig 1,2,3,*, Deyin Lu 1, Amber M. Paul 4, Ram Kuwar 5, Maria Lopez 1, Dobrivoje S. Stokic 3,6 , A. Arturo Leis 6, Michael R. Garrett 7 and Fengwei Bai 1,4 1 Departments of Neurology, University of Mississippi Medical Center, Jackson, MS 39216, USA; [email protected] (D.L.); [email protected] (M.L.); [email protected] (F.B.) 2 Biochemistry, University of Mississippi Medical Center, Jackson, MS 39216, USA 3 Neurobiology & Anatomical Sciences, University of Mississippi Medical Center, Jackson, MS 39216, USA; [email protected] 4 Department of Biological Sciences, University of Southern Mississippi, Hattiesburg, MS 39406, USA; [email protected] 5 Virginia Commonwealth University, Richmond, VA 23284, USA; [email protected] 6 Methodist Rehabilitation Center, Jackson, MS 39216, USA; [email protected] 7 Experimental Therapeutics and Pharmacology, University of Mississippi Medical Center, Jackson, MS 39216, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-601-984-5513; Fax: +1-601-984-6626 Received: 29 October 2018; Accepted: 18 December 2018; Published: 24 December 2018 Abstract: West Nile virus (WNV) infection results in a spectrum of neurological symptoms, ranging from a benign fever to severe WNV neuroinvasive disease with high mortality. Many who recover from WNV neuroinvasive infection present with long-term deficits, including weakness, fatigue, and cognitive problems. While neurons are a main target of WNV, other cell types, especially astrocytes, play an important role in promoting WNV-mediated central nervous system (CNS) damage. -

Delivering Type I Interferon to Dendritic Cells Empowers Tumor Eradication and Immune Combination Treatments
Author Manuscript Published OnlineFirst on November 29, 2017; DOI: 10.1158/0008-5472.CAN-17-1980 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. 1 Delivering type I interferon to dendritic cells empowers tumor eradication and immune combination treatments Anje Cauwels1†, Sandra Van Lint1†, Franciane Paul2, Geneviève Garcin2, Stefaan De Koker1,5, Alexander Van Parys1, Thomas Wueest3, Sarah Gerlo1, José Van der Heyden1, Yann Bordat2, Dominiek Catteeuw1, Elke Rogge1, Annick Verhee1, Bart Vandekerckhove4, Niko Kley3, Gilles Uzé2‡ & Jan Tavernier1,3‡* 1Cytokine Receptor Laboratory, Flanders Institute of Biotechnology, VIB-UGent Center for Medical Biotechnology, Faculty of Medicine and Health Sciences, Ghent University, A. Baertsoenkaai 3, 9000 Ghent, Belgium. 2CNRS UMR 5235, University Montpellier, Place Eugène Bataillon, 34095 Montpellier, France. 3Orionis Biosciences, Technologiepark 4, 9052 Zwijnaarde-Gent, Belgium; and Watertown, Boston, USA. 4Department of Clinical Chemistry, Microbiology and Immunology, Ghent University, UZ Gent, MRB2, De Pintelaan 185, 9000 Gent, Belgium 5Present address: eTheRNA Immunotherapies, Arthur de Coninckstraat 11, 3070 Kortenberg, Belgium *To whom correspondence should be addressed: Jan Tavernier, A. Baertsoenkaai 3, 9000 Ghent, Belgium. Phone +32.9.2649302, Fax +32.9.2649340, [email protected] †shared first authors, ‡shared last authors Downloaded from cancerres.aacrjournals.org on September 28, 2021. © 2017 American Association for Cancer Research. Author Manuscript Published OnlineFirst on November 29, 2017; DOI: 10.1158/0008-5472.CAN-17-1980 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. 2 The authors declare no potential conflicts of interest. Running title: Specific DC-targeted IFN allows nontoxic antitumor efficacy This work was supported by UGent Methusalem and Advanced ERC (CYRE, N° 340941) grants to J.T.; an FWO-V grant G009614N to J.T. -

IL6 Sensitizes Prostate Cancer to the Antiproliferative Effect of Ifna2
H H H Erb et al. IRF9 regulation by IL6 in 20:5 677–689 Research prostate cancer Open Access IL6 sensitizes prostate cancer to the antiproliferative effect of IFNa2 through IRF9 Holger H H Erb1, Regina V Langlechner1, Patrizia L Moser2, Florian Handle1, Tineke Casneuf3, Karin Verstraeten3, Bettina Schlick4, Georg Scha¨fer1, Brett Hall5, Kate Sasser5, Zoran Culig1 and Fre´de´ric R Santer1 Correspondence 1Division of Experimental Urology, Department of Urology 2Department of Pathology, Innsbruck Medical University, should be addressed 6020 Innsbruck, Austria to Z Culig or F R Santer 3Oncology Biomarkers, Janssen Research and Development, Beerse, Belgium Emails 4Oncotyrol, Center for Personalized Medicine, 6020 Innsbruck, Austria [email protected] or 5Oncology Biomarkers, Janssen Research and Development, Spring House, Pennsylvania, USA [email protected] Abstract Development and progression of prostate cancer (PCa) are associated with chronic Key Words inflammation. The cytokine interleukin 6 (IL6) can influence progression, differentiation, " IRF9 survival, and angiogenesis of PCa. To identify novel pathways that are triggered by IL6, we " IL6 performed a gene expression profiling of two PCa cell lines, LNCaP and MDA PCa 2b, treated " prostate cancer with 5 ng/ml IL6. Interferon (IFN) regulatory factor 9 (IRF9) was identified as one of the most " IFNa2 prevalent IL6-regulated genes in both cell lines. IRF9 is a mediator of type I IFN signaling and " inflammation acts together with STAT1 and 2 to activate transcription of IFN-responsive genes. The IL6 Endocrine-Related Cancer regulation of IRF9 was confirmed at mRNA and protein levels by quantitative real-time PCR and western blot respectively in both cell lines and could be blocked by the anti-IL6 antibody Siltuximab. -

The STING-IFN–Dependent Axis Is Markedly Low in Patients With
International Journal of Molecular Sciences Article The STING-IFN-β-Dependent Axis Is Markedly Low in Patients with Relapsing-Remitting Multiple Sclerosis 1, 1, , 1 2 1 Lars Masanneck y, Susann Eichler * y, Anna Vogelsang , Melanie Korsen , Heinz Wiendl , Thomas Budde 3 and Sven G. Meuth 1,2 1 Department of Neurology with Institute of Translational Neurology, University Hospital Münster, 48149 Münster, Germany; [email protected] (L.M.); [email protected] (A.V.); [email protected] (H.W.); [email protected] (S.G.M.) 2 Department of Neurology, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] 3 Institute of Physiology I, University of Münster, 48149 Münster, Germany; [email protected] * Correspondence: [email protected] These authors contributed equally to this work. y Received: 9 November 2020; Accepted: 2 December 2020; Published: 4 December 2020 Abstract: Cyclic GMP-AMP-synthase is a sensor of endogenous nucleic acids, which subsequently elicits a stimulator of interferon genes (STING)-dependent type I interferon (IFN) response defending us against viruses and other intracellular pathogens. This pathway can drive pathological inflammation, as documented for type I interferonopathies. In contrast, specific STING activation and subsequent IFN-β release have shown beneficial effects on experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Although less severe cases of relapse-remitting MS (RRMS) are treated with IFN-β, there is little information correlating aberrant type I IFN signaling and the pathologic conditions of MS. We hypothesized that there is a link between STING activation and the endogenous production of IFN-β during neuroinflammation. -

Monoclonal Antibody to IFNA2 / Interferon Alpha-2 - Aff - Purified
OriGene Technologies, Inc. OriGene Technologies GmbH 9620 Medical Center Drive, Ste 200 Schillerstr. 5 Rockville, MD 20850 32052 Herford UNITED STATES GERMANY Phone: +1-888-267-4436 Phone: +49-5221-34606-0 Fax: +1-301-340-8606 Fax: +49-5221-34606-11 [email protected] [email protected] AM20794PU-S Monoclonal Antibody to IFNA2 / Interferon alpha-2 - Aff - Purified Alternate names: IFN-alpha 2, Interferon alpha-A, LeIF A Quantity: 0.1 mg Concentration: 1.0 mg/ml Background: A member of the type 1 IFN family of related proteins produced by leucocytes and fibroblasts. IFN alpha 1 has antiviral activities. Interferon stimulates the production of two enzymes: a protein kinase and an oligoadenylate synthetase. Uniprot ID: P01563 NCBI: NP_000596.2 GeneID: 3440 Host / Isotype: Mouse / IgG1 Clone: KT5 Immunogen: Recombinant Human IFN-alpha 2a Format: State: Liquid purified Ig fraction Purification: Protein A Chromatography from mouse ascites. Buffer System: PBS containing 0.09% Sodium Azide as a preservative Applications: Indirect ELISA: 1 μg/ml. Other applications not tested. Optimal dilutions are dependent on conditions and should be determined by the user. Specificity: This antibody reacts with both Interferon alpha-2a and Interferon alpha-2b. Species: Human. Other species not tested. Add. Information: CHROMOSOMAL LOCATION Genetic locus: IFNA2 (human) mapping to 9p21.3 Storage: Store the antibody undiluted at 2-8°C for one month or (in aliquots) at -20°C for longer. Avoid repeated freezing and thawing. Shelf life: one year from despatch. General Readings: 1. Adolf, G.R. 1987. Antigenic structure of human interferon-ω1 (interferon-αII1): comparison with other human interferons. -

The MK2 Pathway Is Linked to G-CSF, Cytokine Production and Metastasis
Qeadan et al. J Transl Med (2020) 18:137 https://doi.org/10.1186/s12967-020-02294-z Journal of Translational Medicine RESEARCH Open Access The MK2 pathway is linked to G-CSF, cytokine production and metastasis in gastric cancer: a novel intercorrelation analysis approach Fares Qeadan1, Pranshu Bansal2, Joshua A. Hanson3 and Ellen J. Beswick4* Abstract Background: Gastric cancer is associated with chronic infammation, but there is still much to understand about the tumor microenvironment and the underlying tumor-promoting mechanisms. The Map kinase-activated protein kinase 2 (MK2) pathway is a regulator of infammatory cytokine production that we have been studying in gastrointes- tinal cancers. Here, we set out to determine the signifcance of this gene in gastric cancer along with its downstream mediators and if there were diferences in the primary tumors with and without metastasis. Methods: Human gastric cancer tissues with and without metastasis were examined for MK2 expression and cytokine profle in organ culture supernatants. Advanced statistical methods including a lower triangular correlation matrix, novel rooted correlation network, linear and logistic regression modeling along with Kruskal–Wallis testing with Sidak correction for multiple testing were applied to gain understanding of cytokines/chemokines linked to metastasis. Results: The MK2 pathway is strongly linked with metastasis and a panel of cytokines. Gene expression was able to classify gastric cancer metastasis 85.7% of the time. A signifcant association with a panel of cytokines was found, including G-CSF, GM-CSF, Mip-1β, IFN-α, MCP-1, IL-1β, IL-6, and TNF-α. Mip-1β was found to have the strongest asso- ciation with MK2 and metastasis after Sidak correction for multiple testing. -

Improved Osteogenic Vector for Non-Viral Gene Therapy A.R.A
EuropeanARA Hacobian Cells and et al Materials. Vol. 31 2016 (pages 191-204) DOI: 10.22203/eCM.v031a13 Enhanced ISSN BMP-2 1473-2262 vector IMPROVED OSTEOGENIC VECTOR FOR NON-VIRAL GENE THERAPY A.R.A. Hacobian*, K. Posa-Markaryan, S. Sperger, M. Stainer, D. Hercher, G. A. Feichtinger, C.M.A.P. Schuh and H. Redl Ludwig Boltzmann Institute for Experimental and Clinical Traumatology/AUVA Research Center, The Austrian Cluster for Tissue Regeneration, European Institute of Excellence on Tissue Engineering and Regenerative Medicine Research (Expertissues EEIG), Vienna, Austria. Abstract Introduction Therapeutic compensation of deficient bone regeneration One focus within the field of regenerative medicine is is a challenging task and a topic of on-going search for the reconstruction of bone defects or compensation of novel treatment strategies. One promising approach for deficient regeneration. Several studies have investigated improvement involves non-viral gene delivery using the the practicability of applying growth factors, either in bone morphogenetic protein-2 (BMP-2) gene to provide form of recombinant proteins or by means of gene therapy transient, local and sustained expression of the growth encoding an exogenous gene on a plasmid (Feichtinger factor. However, since efficiency of non-viral gene delivery et al., 2014; Kempen et al., 2010; Koh et al., 2008; Sood is low, this study focused on the improvement of a BMP- et al., 2012). Recombinant proteins used in therapeutic 2 gene expression system, aiming for compensation of approaches generally show a short half-life at the target site poor transfection efficiency. First, the native BMP-2 gene (Alt et al., 2009; Sandhu, 2004). -
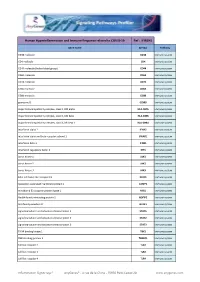
Hyperinflammation& Immune Response.Xlsx
Human Hyperinflammation and Immune Response related to COVID-19 Ref : IFIR1H1 Gene Name Symbol Pathway CD38 molecule CD38 Immune system CD4 molecule CD4 Immune system CD44 molecule (Indian blood group) CD44 Immune system CD69 molecule CD69 Immune system CD74 molecule CD74 Immune system CD8a molecule CD8A Immune system CD8b molecule CD8B Immune system granzyme B GZMB Immune system major histocompatibility complex, class II, DM alpha HLA-DMA Immune system major histocompatibility complex, class II, DM beta HLA-DMB Immune system major histocompatibility complex, class II, DR beta 1 HLA-DRB1 Immune system interferon alpha 2 IFNA2 Immune system interferon alpha and beta receptor subunit 1 IFNAR1 Immune system interferon beta 1 IFNB1 Immune system interferon regulatory factor 3 IRF3 Immune system Janus kinase 1 JAK1 Immune system Janus kinase 2 JAK2 Immune system Janus kinase 3 JAK3 Immune system killer cell lectin like receptor C1 KLRC1 Immune system lysosomal associated membrane protein 1 LAMP1 Immune system mindbomb E3 ubiquitin protein ligase 1 MIB1 Immune system Nedd4 family interacting protein 2 NDFIP2 Immune system NLR family member X1 NLRX1 Immune system signal transducer and activator of transcription 1 STAT1 Immune system signal transducer and activator of transcription 2 STAT2 Immune system signal transducer and activator of transcription 3 STAT3 Immune system TANK binding kinase 1 TBK1 Immune system TBK1 binding protein 1 TBKBP1 Immune system toll like receptor 2 TLR2 Immune system toll like receptor 3 TLR3 Immune system toll like -

Structural and Dynamic Determinants of Type I Interferon Receptor
Jacob Piehler Structural and dynamic Christoph Thomas determinants of type I interferon K. Christopher Garcia Gideon Schreiber receptor assembly and their functional interpretation Authors’ addresses Summary: Type I interferons (IFNs) form a network of homologous Jacob Piehler1,*, Christoph Thomas2, K. Christopher Garcia2, cytokines that bind to a shared, heterodimeric cell surface receptor and Gideon Schreiber3,* engage signaling pathways that activate innate and adaptive immune 1Department of Biology, University of Osnabru¨ck, responses. The ability of IFNs to mediate differential responses through Osnabru¨ck, Germany. the same cell surface receptor has been subject of a controversial debate 2Departments of Molecular and Cellular Physiology, and and has important medical implications. During the past decade, a Structural Biology, Howard Hughes Medical Institute, comprehensive insight into the structure, energetics, and dynamics of Stanford University School of Medicine, Stanford, CA, USA. IFN recognition by its two-receptor subunits, as well as detailed corre- 3Department of Biological Chemistry, Weizmann Institute lations with their functional properties on the level of signal activation, of Science, Rehovot, Israel. gene expression, and biological responses were obtained. All type I IFNs bind the two-receptor subunits at the same sites and form struc- *Jacob Piehler and Gideon Schreiber contributed equally as turally very similar ternary complexes. Differential IFN activities were senior authors. found to be determined by different lifetimes and ligand affinities toward the receptor subunits, which dictate assembly and dynamics of Correspondence to: the signaling complex in the plasma membrane. We present a simple Gideon Schreiber model, which explains differential IFN activities based on rapid endo- Department of Biological Chemistry cytosis of signaling complexes and negative feedback mechanisms Weizmann Institute of Science interfering with ternary complex assembly. -

The NASA Twins Study
RESEARCH ◥ state within the time period of the study. These RESEARCH ARTICLE SUMMARY included changes in telomere length, gene regulation measured in both epigenetic and transcriptional data, gut microbiome compo- HUMAN PHYSIOLOGY sition, body weight, carotid artery dimensions, subfoveal choroidal thickness and peripapillary total retinal thickness, and serum metabolites. The NASA Twins Study: In addition, some factors were significantly af- fected by the stress of re- ◥ A multidimensional analysis of a ON OUR WEBSITE turning to Earth, including Read the full article inflammation cytokines year-long human spaceflight at http://dx.doi. and immune response gene org/10.1126/ networks,aswellascog- science.aau8650 nitive performance. For Francine E. Garrett-Bakelman et al. .................................................. a few measures, persistent changes were observed even after 6 months on INTRODUCTION: To date, 559 humans have microbial shifts, and alterations in gene reg- Earth, including some genes’ expression levels, been flown into space, but long-duration ulation. However, previous studies collected increased DNA damage from chromosomal in- (>300 days) missions are rare (n = 8 total). very limited data, did not integrate simulta- versions, increased numbers of short telomeres, Long-duration missions that will take humans neous effects on multiple systems and data and attenuated cognitive function. to Mars and beyond are planned by public types in the same subject, or were restricted to Downloaded from and private entities for the 2020s and 2030s; 6-month missions. Measurement of the same CONCLUSION: Given that the majority of the therefore, comprehensive studies are needed variables in an astronaut on a year-long mis- biological and human health variables remained now to assess the impact of long-duration sion and in his Earth-bound twin indicated stable, or returned to baseline, after a 340-day spaceflight on the human body, brain, and the biological measures that might be used to space mission, these data suggest that human overall physiology.