Renal Manifestations of Common Variable Immunodeficiency Tiffany
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spontaneous Reversal of Acquired Autoimmune Dysfibrinogenemia Probably Due to an Antiidiotypic Antibody Directed to an Interspec
Spontaneous reversal of acquired autoimmune dysfibrinogenemia probably due to an antiidiotypic antibody directed to an interspecies cross-reactive idiotype expressed on antifibrinogen antibodies. A Ruiz-Arguelles J Clin Invest. 1988;82(3):958-963. https://doi.org/10.1172/JCI113704. Research Article A young man with a long history of abnormal bleeding was seen in January 1985. Coagulation tests showed dysfibrinogenemia and an antifibrinogen autoantibody was demonstrable in his serum. This antibody, when purified, was capable of inhibiting the polymerization of normal fibrin monomers, apparently through binding to the alpha fibrinogen chain. 6 mo later the patient was asymptomatic, coagulation tests were normal, and the antifibrinogen autoantibody was barely detectable. At this time, affinity-purified autologous and rabbit antifibrinogen antibodies were capable of absorbing an IgG kappa antibody from the patient's serum, which reacted indistinctly with both autologous and xenogeneic antifibrinogen antibodies in enzyme immunoassays. It has been concluded that the patient's dysfibrinogenemia was the result of an antifibrinogen autoantibody, and that later on an anti-idiotype antibody, which binds an interspecies cross- reactive idiotype expressed on anti-human fibrinogen antibodies, inhibited the production of the antifibrinogen autoantibody which led to the remission of the disorder. Find the latest version: https://jci.me/113704/pdf Spontaneous Reversal of Acquired Autoimmune Dysfibrinogenemia Probably Due to an Antildiotypic Antibody Directed to an Interspecies Cross-reactive Idiotype Expressed on Antifibrinogen Antibodies Alejandro Ruiz-Arguelles Department ofImmunology, Laboratorios Clinicos de Puebla, Puebla, Puebla 72530, Mexico Abstract disorder. This anti-Id antibody was shown to react with xeno- geneic antifibrinogen antibodies, hence, its specificity is an A young man with a long history of abnormal bleeding was interspecies cross-reactive Id (IdX)' most likely encoded by seen in January 1985. -

Diagnosis and Primary Care Management of Focal Segmental Glomerulosclerosis in Children
1.5 CONTACTCOCONOONNTATACACACT HOURSHOUROURRS 1.5 CONTACT HOURS Monkey Business Images / Thinkstock Diagnosis and primary care management of focal segmental glomerulosclerosis in children Abstract: Focal segmental glomerulosclerosis (FSGS) is a pattern of kidney damage that can occur in individuals at any age, including children. Pediatric patients with FSGS require medication monitoring, growth, and psychological health. This article discusses the NP’s role in the clinical presentation, diagnostic workup, and treatment of FSGS in pediatric patients. By Angela Y. Wong, MS, CPNP-PC and Rita Marie John, DNP, EdD, CPNP-PC, FAANP P, a 10-year-old girl, told her parents several This article discusses the pathophysiology, epide- times, “I feel puffy,” before the family sought miology, clinical presentation, and treatment options J medical attention. This complaint could in- for FSGS. In addition, the article elucidates the role dicate a myriad of issues ranging from sudden weight of the NP in managing this disease when it occurs gain to simple abdominal gas. For this child, “puffy” during childhood. depicted her progressive edema—the only overt symp- tom of her diagnosis of focal segmental glomerulo- ■ Overview sclerosis (FSGS). Although some children with FSGS FSGS is a pattern of kidney damage involving scarring may only present with asymptomatic proteinuria at of glomeruli in the kidney. This pattern of glomerulo- a routine physical, FSGS can affect individuals of all sclerosis is focal and segmental, meaning not all glom- ages, and is a common cause of end-stage renal disease eruli are affected and only parts of the glomeruli are (ESRD) in children.1 damaged, respectively.2 FSGS is the most common Keywords: end-stage renal disease, focal segmental glomerulosclerosis, kidney transplant, nephrotic syndrome, pediatrics 28 The Nurse Practitioner • Vol. -

Guidelines for Writing Examination Items (Questions)
Guidelines for Writing Examination Items (Questions) Enclosed are the content outlines for the Immunology certification examinations. The content outline specifies the breakdown of content and overall structure of the examination and indicates how many test questions are assigned to each topic area from a total of 70 questions. The content outline will guide you in creating new items to match certain topic areas. We would appreciate at least two (2) new items for each major roman numeral on the content outline(s). This means we are asking you to write two items for Roman numeral I, two items for Roman numeral II, and so on for each of the Roman numeral sections of the Content Outline. We would appreciate items submitted in advance, preferably no later than Tuesday, February 25, 2020. • Please use the “Item Writing Template” to create your new items. This is the proper format to be used for all items you submit. Font: Times New Roma. Font Size: 11 • Please identify the Content Outline position for each item (e.g., Chemistry Content Outline, Roman numeral I. “Proteins”, A. “Total Proteins” should be noted as “I.A.”). • New items must be multiple choice with four (4) possible answers. Remember to avoid "double negatives," "not" questions (e.g., "Which of the following is NOT true?"), and those allowing "all (or none) of the above," or "a and b" as a possible answer. • Each new item that you create must be accompanied with a reference [Author, Publication Year, Title, Edition, Page Number(s)] containing the correct answer. IMPORTANT: references must be from an AAB Review Manual, Governmental Regulations, Association or World Health Organization (WHO) Guidelines, or a text or manual published within the last six (6) years. -

Glomerulosclerosis in Reflux Nephropathy
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Kidney International, Vol. 21(1982), pp. 528—534 NEPHROLOGY FORUM Glomeruloscierosis in reflux nephropathy Principal discussant: RAMzI S. COTRAN Department of Pathology, Brigham and Women's Hospital, Boston, Massachusetts penis, testes, and urethral meatus were normal. The prostate was of normal size. Editors The BUN was 14 mg/dl; creatinine, 1.3 mg/dl (creatinine clearance, 113 mI/mm); blood chemistries were normal; the blood glucose was 93 JORDANJ. COHEN mg/dl; and the complement profile was normal. Serum protein was 7.5 JOHN 1. HARRINGTON gIdI with 4.3 g/dl albumin. Urinalysis showed a pH of 5; a specific JEROME P.KASSIRER gravity of 1.015; 4+ protein, no cells, and no bacteria. Urine culture was sterile. The 24-hour urine protein excretion was 2.4 g. Editor Chest x-ray showed borderline cardiomegaly with clear lungs. An Managing intravenous pyelogram revealed bilateral coarse scarring with caliecta- CHERYL J. ZUSMAN sis. The right kidney was smaller than the left. There was moderate ureterectasia extending down to the ureterovesical junction. A voiding cystourethrogram revealed a large-capacity bladder; the patient had no MichaelReese Hospital and Medical Center urge to void after almost 500 ml of contrast material was instilled. Bilateral reflux was greater and persistent on the left and was intermit- University of Chicago, tent on the right. The left ureter was dilated and tortuous. A left Pritzker School of Medicine ureterocele and right bladder diverticulum were visualized. and A biopsy of the left kidney showed focal scarring with interstitial New England Medical Center fibrosis and chronic inflammation, tubular atrophy, and dilation. -

Focal Segmental Glomerulosclerosis in Systemic Lupus Erythematosus
Relato de Caso Focal Segmental Glomerulosclerosis in Systemic Lupus Erythematosus: One or Two Glomerular Diseases? Glomerulosclerose Segmentar e Focal em Lúpus Eritematoso Sistêmico: Uma ou Duas Doenças? Rogério Barbosa de Deus1, Luis Antonio Moura1, Marcello Fabiano Franco2, Gianna Mastroianni Kirsztajn1 1 Nephrology Division and 2 Department of Pathology - Universidade Federal de São Paulo – EPM/UNIFESP- São Paulo. ABSTRACT Some patients with clinical and/or laboratory diagnosis of systemic lupus erythematosus (SLE) present with nephritis which from the morphological point of view does not fit in one of the 6 classes described in the WHO classification of lupus nephritis. On the other hand, nonlupus nephritis in patients with confirmed SLE is rarely reported. This condition may not be so uncommon as it seems. The associated glomerular lesions most frequently described are amyloidosis and focal segmental glomerulosclerosis (FSGS). We report on a 46 year-old, caucasian woman, who fulfilled the American College of Rheumatology criteria for SLE diagnosis: arthritis, positive anti-DNA, ANA, anti-Sm antibodies, and cutaneous maculae. During the follow-up, she presented arthralgias, alopecia, vasculitis, lower extremities edema and decreased serum levels of C3 and C4. Proteinuria was initially nephrotic, but reached negative levels. The serum creatinine varied from 0.7 to 3.0 mg/dl. The patient was submitted to the first renal biopsy at admission and to the second one, 3 years later, with diagnosis of minimal change disease and FSGS, respectively. No deposits were demonstrated by immunofluorescence. In the present case, we believe that the patient had SLE and developed an idiopathic disease of the minimal change disease-FSGS spectrum. -

El Paso Community College Syllabus Part II Official Course Description
MLAB 1235; Revised Fall 2019/Spring 2020 El Paso Community College Syllabus Part II Official Course Description SUBJECT AREA Medical Laboratory Technology COURSE RUBRIC AND NUMBER MLAB 1235 COURSE TITLE Immunology/Serology COURSE CREDIT HOURS 2 1 : 3 Credits Lec Lab I. Catalog Description Provides an introduction to the theory and application of basic immunology, including the immune response, principles of antigen-antibody reactions, and the principles of serological procedures as well as quality control, quality assurance, and lab safety. A grade of “C” or better is required in this course to take the next course. Corequisite: MLAB 1260. (1:3). Lab fee. II. Course Objectives A. Unit I. Laboratory Operations Upon satisfactory completion of this unit, the student will be able to: 1. Demonstrate adherence to Standard Precautions and the organizations’ SOP (Standard Operating Procedures) at all times. 2. Discuss legal and ethical concerns pertaining to Patient Informed Consent, Standard of Care, and HIPAA regulations. 3. Compliance with government, state, and organizational safety regulations involving Biological, Chemical, Radioactive, Fire, Physical, and Electrical hazards. 4. Explain the importance of actively participating in Quality Assurance, Quality Control and Proficiency Testing protocols incorporating precision, accuracy, Levi Jennings Charts and Westgard Rules. 5. Locate and make use of MSDS (Material Safety Data Sheets) 6. Discuss how OSHA affects safety, health, and compliance policies in the workplace. 7. Discuss nosocomial infections and identify the basic programs for infection control. 8. Identify the potential routes of infection and methods for preventing transmission of microorganisms through these routes. 9. Explain the proper techniques for hand washing, gowning, gloving, and masking. -

A Challenging Case of Igd Kappa Multiple Myeloma Associated with Primary Amyloidosis: Importance of Serum Free Light Chains in M
L al of euk rn em u i o a J Journal of Leukemia García de Veas Silva JL et al, J Leuk 2014, 2:5 ISSN: 2329-6917 DOI: 10.4172/2329-6917.1000164 Case Report Open Access A Challenging Case of IgD Kappa Multiple Myeloma Associated With Primary Amyloidosis: Importance of Serum Free Light Chains in Monitoring Treatment Response and Disease Relapse José Luis García de Veas Silva1*, Carmen Bermudo Guitarte1, Paloma Menéndez Valladares1, Rafael Duro Millán2 and Johanna Carolina Rojas Noboa2 1Department of Clinical Biochemistry, Hospital Universitario Virgen Macarena, Sevilla, Spain 2Department of Hematology, Hospital Universitario Virgen Macarena, Sevilla, Spain *Corresponding author: José Luis García de Veas Silva, Laboratory of Proteins, Department of Clinical Biochemistry, Hospital Universitario Virgen Macarena, Sevilla, Spain, Tel: +034955008108; E-mail: [email protected] Rec date: Oct 10, 2014, Acc date: Oct 16, 2014; Pub date: Oct 24, 2014 Copyright: © 2014 García de Veas Silva JL, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Multiple Myeloma (MM) is a malignancy of B cells characterized by an atypical proliferation of plasma cells. IgD MM has a very low incidence (2% of total MM cases) and it´s characterized by an aggressive course and a worse prognosis than other subtypes. The serum free light chains (sFLC) are very important markers for monitoring patients with MM and other monoclonal gammopathies. When the sFLC are present in low concentrations, it is often difficult to detect them by conventional methods such as serum protein electrophoresis and serum immunofixation. -

Diagnosis and Management of Pyelonephritis
DIAGNOSIS AND MANAGEMENT OF PYELONEPHRITIS ROBERT D. TAYLOR, M.D. Research Division F the four common nephropathies, glomerulonephritis, nephrosclerosis, Ointercapillary glomerulosclerosis and pyelonephritis, at present only pyelonephritis, the most familiar of these, offers the possibility of arrest or cure. It has been strangely neglected, possibly because it is a nosologic step- child, half surgical and half medical. Volhard and Fahr in 19141 did not list it among medical diseases of the kidney nor did Addis in 1925.2 Apart from the pediatric, obstetric and urologic aspects, little of general interest was written about it until 1939 when Weiss and Parker3 explored its relationship to hyper- icnsive disease. In 1948 Raaschau4 of Copenhagen published an excellent monograph on the subject with extensive pathologic and clinical studies. His most striking observation concerns its incidence. Among 3607 routine autop- sies he found histopathologic evidence of pyelonephritis in 5.6 per cent. One half of these persons had died of renal failure but in only 1 out of 6 was the condition recognized ante mortem. Undoubtedly, if a disease is both common and remediable, its diagnosis and treatment are of general interest. Etiology and Pathology The most common organisms in pyelonephritis are colon bacilli and hemo- lytic streptococci. In some cases the infection is secondary to lesions which prevent free urinary drainage. In this group surgical correction of the causes of urinary stasis usually permits eradication of infection. Among the remaining patients the process is primary with no demonstrable anatomic abnormality. The organisms apparently reach the kidney substance through the blood stream. The pelvis and lower urinary tract become involved by contamination with infected urine. -

Focal Segmental Glomerulosclerosis
some = Focal sections of = Segmental Focal Segmental kidney filters = Glomerulo FSGS Glomerulosclerosis = { are scarred Sclerosis How can research help? Research, which often requires patient participation, can lead to a better understanding of the causes, provide better diagnoses and more effective treatments, and ultimately help to find a cure. Be part of the discovery: By studying patients, researchers can more quickly unlock the mysteries of this disease. To get involved in research and stay informed, go to: Nephrotic Syndrome Study Network www.Nephrotic-Syndrome- NEPTUNE is a part of the NIH Rare Diseases Clinical Research Network (RDCRN). Funding and/or programmatic support for this project has been provided by U54 Studies.org DK083912 from the NIDDK and the NIH Office of Rare Diseases Research (ORDR), the NephCure Foundation and the University of Michigan. The views expressed in written materials of publications do not necessarily reflect the official policies of the or call 1-866-NephCure Department of Health and Human Services; nor does mention by trade names, commercial practices or organizations imply endorsement by the U.S. Government. What is Focal and Segmental Glomerulosclerosis? Symptoms, Diagnosis and Treatment Each person has Focal Segmental Glomerulosclerosis (FSGS) is a Your nephrologist may also recommend: two kidneys in rare disease that attacks the kidney’s filtering system • Diuretics and low salt diet help to control edema their lower back. (glomeruli) causing scarring of the filter. FSGS • A medication that blocks a hormone system called is one of the causes of a condition known as the renin angiotensin system (ACE inhibitor or ARB) Nephrotic Syndrome (NS). -
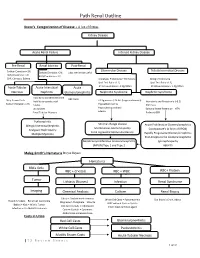
Path Renal Outline
Path Renal Outline Krane’s Categorization of Disease + A lot of Extras Kidney Disease Acute Renal Failure Intrinsic Kidney Disease Pre‐Renal Renal Intrinsic Post‐Renal Sodium Excretion <1% Glomerular Disease Tubulointerstitial Disease Sodium Excretion < 1% Sodium Excretion >2% Labs aren’t that useful BUN/Creatinine > 20 BUN/Creatinine < 10 CHF, Cirrhosis, Edema Urinalysis: Proteinuria + Hematuria Benign Proteinuria Spot Test Ratio >1.5, Spot Test Ratio <1.5, Acute Tubular Acute Interstitial Acute 24 Urine contains > 2.0g/24hrs 24 Urine contains < 1.0g/24hrs Necrosis Nephritis Glomerulonephritis Nephrotic Syndrome Nephritic Syndrome Inability to concentrate Urine RBC Casts Dirty Brown Casts Inability to secrete acid >3.5g protein / 24 hrs (huge proteinuria) Hematuria and Proteinuria (<3.5) Sodium Excretion >2% Edema Hypoalbuminemia RBC Casts Hypercholesterolemia Leukocytes Salt and Water Retention = HTN Focal Tubular Necrosis Edema Reduced GFR Pyelonephritis Minimal change disease Allergic Interstitial Nephritis Acute Proliferative Glomerulonephritis Membranous Glomerulopathy Analgesic Nephropathy Goodpasture’s (a form of RPGN) Focal segmental Glomerulosclerosis Rapidly Progressive Glomerulonephritis Multiple Myeloma Post‐Streptococcal Glomerulonephritis Membranoproliferative Glomerulonephritis IgA nephropathy (MPGN) Type 1 and Type 2 Alport’s Meleg‐Smith’s Hematuria Break Down Hematuria RBCs Only RBC + Crystals RBC + WBC RBC+ Protein Tumor Lithiasis (Stones) Infection Renal Syndrome Imaging Chemical Analysis Culture Renal Biopsy Calcium -

FSGS Fact Sheet 4.12.18
® Focal Segmental Glomerulosclerosis Focal Segmental Glomerulosclerosis (FSGS) is a rare kidney disease characterized by dysfunction in the part of the kidney that filters blood (glomeruli). Only some glomeruli are aected, but continued damage can lead to kidney failure. Focal = Some Segmental = Sections Glomerulo = of the Filtering Units Sclerosis = Are Scarred FSGS Symptoms The exact cause of primary Early symptoms of FSGS are the same as Nephrotic Syndrome. FSGS is unknown and not precisely understood. However, genetic and Common Symptoms: environmental factors may be associated - Protein in the urine, which can be foamy (called with the disease. proteinuria) - Low levels of protein in the blood With FSGS, many individuals - Swelling in parts of the body, most noticeably around experience cycles the eyes, hands, feet, and abdomen (called edema) of remission and relapse. - Weight gain due to extra fluid building up in your body Remission means there - Can cause high blood pressure (called hypertension) is currently no and high fat levels in the blood (high cholesterol) protein spilling into the 50% of urine. patients with FSGS will Fast Facts progress to kidney failure. The only way to dierentiate FSGS from other primary Every FSGS Nephrotic Syndrome conditions is to have a kidney biopsy. patient follows a unique journey. FSGS in Adults FSGS in Children - FSGS occurs more - Focal Segmental frequently in adults than Glomerulosclerosis is one of in children and is most the leading causes of End Some patients prevalent in adults 45 Stage Renal Disease receive a kidney years or older. (ESRD) in children. transplant to treat their kidney failure due to FSGS, - African Americans are 5 - FSGS is associated with but FSGS comes back times more likely to get up to 20% of all new cases to attack the new FSGS in comparison with of Nephrotic Syndrome kidney 30-50% the general population. -

Monitoring Multiple Myeloma Patients Treated with Daratumumab: Teasing out Monoclonal Antibody Interference
Clin Chem Lab Med 2016; 54(6): 1095–1104 Open Access Christopher McCuddena, Amy E. Axela, Dominique Slaets, Thomas Dejoie, Pamela L. Clemens, Sandy Frans, Jaime Bald, Torben Plesner, Joannes F.M. Jacobs, Niels W.C.J. van de Donk, Philippe Moreau, Jordan M. Schecter, Tahamtan Ahmadi and A. Kate Sasser* Monitoring multiple myeloma patients treated with daratumumab: teasing out monoclonal antibody interference DOI 10.1515/cclm-2015-1031 developed using a mouse anti-daratumumab antibody. To Received October 21, 2015; accepted February 10, 2016; previously evaluate whether anti-daratumumab bound to and shifted published online March 30, 2016 the migration pattern of daratumumab, it was spiked into Abstract daratumumab-containing serum and resolved by IFE/SPE. The presence (DIRA positive) or absence (DIRA negative) Background: Monoclonal antibodies are promising anti- of residual M-protein in daratumumab-treated patient myeloma treatments. As immunoglobulins, monoclonal samples was evaluated using predetermined assessment antibodies have the potential to be identified by serum criteria. DIRA was evaluated for specificity, limit of sensi- protein electrophoresis (SPE) and immunofixation elec- tivity, and reproducibility. trophoresis (IFE). Therapeutic antibody interference with Results: In all of the tested samples, DIRA distinguished standard clinical SPE and IFE can confound the use of between daratumumab and residual M-protein in com- these tests for response assessment in clinical trials and mercial serum samples spiked with daratumumab and disease monitoring. in daratumumab-treated patient samples. The DIRA Methods: To discriminate between endogenous myeloma limit of sensitivity was 0.2 g/L daratumumab, using protein and daratumumab, a daratumumab-specific spiking experiments. Results from DIRA were repro- immunofixation electrophoresis reflex assay (DIRA) was ducible over multiple days, operators, and assays.