Analysis of 52 Pesticides in Fresh Fish Muscle by Quechers Extraction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pesticide Action Networknews
30th Anniversary Edition Pesticide Action Network NEWS Advancing alternatives to pesticides worldwide • www.panna.org Year-end 2012 Cultivating the roots of health and justice Pesticide Action Network: The First 30 Years By 1982, the luster of industrial agriculture—the so-called “Green Revolution”—had faded in developing countries. The promised dramatic increases in yields from “miracle” hybrid grains that required high inputs of water, chemical fertilizers and pesticides failed to deliver and were revealed as campaigns to sell technology to people who couldn’t afford it. Local communities were losing control over their own food systems, and women and children shouldered more of the fieldwork—and bore the brunt of pesticide exposure. The global pesticide trade was, however, yielding dramatic profits for chemical companies as more and more farmers were trapped on a pesticide treadmill. That was the world when PAN was founded. In the years since, the world community has reassessed. When rice farming was collapsing in the 1980s due to pest resurgence from resistance to pesticides, Indonesia needed alternatives. A com- ifty years after Silent Spring and 30 years bination of community-scale peer-learning projects recaptured Fafter PAN’s founding, our struggle for health Indigenous farming knowledge and wove it into new ecological pest management. “Farmer Field Schools”—today adapted to and justice remains vital and more urgent than local needs in many countries—returned bountiful crops of rice ever. Challenging the global proliferation of while expenditures on agrichemicals were slashed. By 2002, more pesticides is about challenging corporate control, than a million Indonesian farmers had participated in field schools that became models for localized sustainable agriculture in other ensuring scientific integrity and defending basic countries. -

Complaint for Declaratory and Injunctive Relief 1 1 2 3 4 5 6 7 8 9
1 Justin Augustine (CA Bar No. 235561) Jaclyn Lopez (CA Bar No. 258589) 2 Center for Biological Diversity 351 California Street, Suite 600 3 San Francisco, CA 94104 Tel: (415) 436-9682 4 Fax: (415) 436-9683 [email protected] 5 [email protected] 6 Collette L. Adkins Giese (MN Bar No. 035059X)* Center for Biological Diversity 8640 Coral Sea Street Northeast 7 Minneapolis, MN 55449-5600 Tel: (651) 955-3821 8 Fax: (415) 436-9683 [email protected] 9 Michael W. Graf (CA Bar No. 136172) 10 Law Offices 227 Behrens Street 11 El Cerrito, CA 94530 Tel: (510) 525-7222 12 Fax: (510) 525-1208 [email protected] 13 Attorneys for Plaintiffs Center for Biological Diversity and 14 Pesticide Action Network North America *Seeking admission pro hac vice 15 16 IN THE UNITED STATES DISTRICT COURT 17 FOR THE NORTHERN DISTRICT OF CALIFORNIA 18 SAN FRANCISCO DIVISION 19 20 CENTER FOR BIOLOGICAL ) 21 DIVERSITY, a non-profit organization; and ) Case No.__________________ PESTICIDE ACTION NETWORK ) 22 NORTH AMERICA, a non-profit ) organization; ) 23 ) Plaintiffs, ) COMPLAINT FOR DECLARATORY 24 ) AND INJUNCTIVE RELIEF v. ) 25 ) ENVIRONMENTAL PROTECTION ) 26 AGENCY; and LISA JACKSON, ) Administrator, U.S. EPA; ) 27 ) Defendants. ) 28 _____________________________________ ) Complaint for Declaratory and Injunctive Relief 1 1 INTRODUCTION 2 1. This action challenges the failure of Defendants Environmental Protection Agency and 3 Lisa Jackson, Environmental Protection Agency Administrator, (collectively “EPA”) to consult with the 4 United States Fish and Wildlife Service (“FWS”) and National Marine Fisheries Service (“NMFS”) 5 (collectively “Service”) pursuant to Section 7(a)(2) of the Endangered Species Act (“ESA”), 16 U.S.C. -

Sound Management of Pesticides and Diagnosis and Treatment Of
* Revision of the“IPCS - Multilevel Course on the Safe Use of Pesticides and on the Diagnosis and Treatment of Presticide Poisoning, 1994” © World Health Organization 2006 All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by the World Health Organization to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either expressed or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use. CONTENTS Preface Acknowledgement Part I. Overview 1. Introduction 1.1 Background 1.2 Objectives 2. Overview of the resource tool 2.1 Moduledescription 2.2 Training levels 2.3 Visual aids 2.4 Informationsources 3. Using the resource tool 3.1 Introduction 3.2 Training trainers 3.2.1 Organizational aspects 3.2.2 Coordinator’s preparation 3.2.3 Selection of participants 3.2.4 Before training trainers 3.2.5 Specimen module 3.3 Trainers 3.3.1 Trainer preparation 3.3.2 Selection of participants 3.3.3 Organizational aspects 3.3.4 Before a course 4. -

Determination of Selected Priority Pesticides in High Water Fruits and Vegetables by Modified Quechers and GC-ECD with GC-MS/MS Confirmation
molecules Article Determination of Selected Priority Pesticides in High Water Fruits and Vegetables by Modified QuEChERS and GC-ECD with GC-MS/MS Confirmation Maciej Tankiewicz Department of Environmental Toxicology, Faculty of Health Sciences with Subfaculty of Nursing and Institute of Maritime and Tropical Medicine, Medical University of Gda´nsk,D˛ebowaStr. 23A, 80-204 Gda´nsk,Poland; [email protected]; Tel.: +48-58-349-1937 Received: 21 December 2018; Accepted: 23 January 2019; Published: 24 January 2019 Abstract: A modified quick, easy, cheap, efficient, rugged and safe (QuEChERS) method coupled to gas chromatography with electron capture detector (GC-ECD) was developed for simultaneous determination of selected electronegative pesticides in fruits and vegetables with high water content. The chosen compounds are commonly detected in fruit and vegetable crops, and some of their metabolites have even been found in human urine. In addition, some of them are known or suspected carcinogens according to the International Agency for Research of Cancer. Extraction and clean up parameters were optimized, thus the original QuEChERS method was modified to decrease solvent usage, in accordance with ‘green chemistry’ principles. The proposed methodology was validated in terms of selectivity, specificity, linearity, precision and accuracy. The obtained limits of detection (LODs) for all investigated pesticides ranged from 5.6 µg·kg−1 to 15 µg·kg−1 and limits of quantification (LOQs) from 17 µg·kg−1 to 45 µg·kg−1. The obtained data demonstrated the good reproducibility and stability of the procedure in the tested concentration range up to 10 mg·kg−1, with relative standard deviations (RSDs) lower than 10%. -

4C Pesticide Lists
4C PESTICIDE LISTS Version 4.0 II 4C PESTICIDE LISTS Copyright notice © 2020 4C Services GmbH This document is protected by copyright. It is freely available from the 4C website or upon request. No part of this copyrighted document may be changed or amended. The document may not be duplicated or copied in any form or by any means for commercial purpose without permission of 4C Services. Document Title: 4C Pesticide Lists Version 4.0 Valid from: 01 July 2020 III Content List of Tables ........................................................................................................................ IV Abbreviations ....................................................................................................................... IV 4C PESTICIDE LISTS 1 Introduction ................................................................................................................... 5 2 Selection Criteria Used for the 4C Pesticide Lists .......................................................... 5 3 4C Red List Pesticides: 4C Code of Conduct Requirements and Actions to be Promoted .................................. 6 4 4C Yellow List Pesticides: 4C Code of Conduct Requirements and Actions to be Promoted .................................. 7 © 4C Services GmbH IV List of Tables Table 1: 4C list of unacceptable pesticides ............................................................................ 8 Table 2: 4C red pesticide list ................................................................................................. 9 Table -
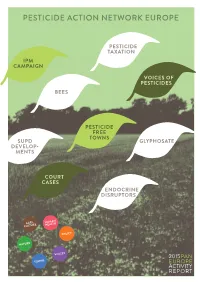
Pesticide Action Network Europe
PESTICIDE ACTION NETWORK EUROPE PESTICIDE TAXATION IPM CAMPAIGN VOICES OF PESTICIDES BEES PESTICIDE FREE TOWNS SUPD GLYPHOSATE DEVELOP- MENTS COURT CASES ENDOCRINE DISRUPTORS AGRI- HUMAN CULTURE HEALTH POLICY NATURE VOICES 2015PAN TOWNS EUROPE ACTIVITY REPORT WHO WE ARE 3 WHAT WE DO 5 PAN EUROPE & THE SUSTAINABLE USE DIRECTIVE 9 INTEGRATED PEST MANAGEMENT 11 PESTICIDE FREE TOWNs 13 BEEs 15 ENDOCRINE DISRUPTING CHEMICALs 17 FURTHER THREATS (GLYPHOSATE, ETC.) 19 COURT CASEs 21 VAT & PESTICIDE TAXATION 23 PESTICIDE SALE 2011-2013 – BASELINE YEAR 2011=100 TONS OF PESTICIDES SOLD IN 2011–2013 WHO WE ARE Pesticide Action Network (PAN) was founded in 1982 and is a network of over 600 non- governmental organisations, institutions and individuals in over 60 countries worldwide working to minimise the negative effects and replace the use of harmful pesticides with eco- logically sound alternatives. Its projects and campaigns are coordinated by five autonomous Regional Centres. PAN Europe is the regional centre in Europe. Located in Brussels, it was founded in 1987 and brings together 34 consumer, public health, and environmental organisations, trades unions, women’s groups and farmer associations from across 21 European countries. PAN Europe’s vision is of a world in which high agricultural productivity is achieved by truly sustainable agricultural production systems in which agrochemical inputs and environmental damage are minimised, and where local people control local production using local varieties. WHY THE FIGHT ON PESTICIDES IS IMPORTANT All of us are exposed directly or indirectly to pesticides and other agrochemicals- farm workers and their families most of all, but every consumer will be exposed to dozens of different pesti- cides every day through food and the environment. -

Acknowledgements
Acknowledgements We would like to thank those involved in creating Planning a Drift Catcher Project and Organizing a Drift Campaign, including: Jeff Conant from the Hesperian Foundation; Mateo Rutherford and Roy Rojas of BITTS for translation; Brenda J. Willoughby (Pesticide Action Network North America) for layout; and contributors Andrea Wilson and Tracey Brieger (Californians for Pesticide Reform) and Katherine Mills, Susan Kegley, Tanya Brown, Kelly Campbell and Christine Riordan (Pesticide Action Network North America). Major funding for this guide and development of the Drift Catcher was provided by the Cedar Tree Foundation. Additional support was provided by grants to Pesticide Action Network North America and/or Californians for Pesticide Reform by the Beldon Fund, The California Endowment, The California Wellness Foundation, Columbia Foundation, Nathan Cummings Foundation, David B. Gold Foundation, Richard and Rhoda Goldman Foundation, Clarence E. Heller Charitable Foundation, David H. Klein, Jr. Foundation and John Merck Fund. The authors bear responsibility for any factual errors. Recommendations and views expressed are those of Pesticide Action Network North America, and do not necessarily represent the views of our funders and supporters. © 2012 by Pesticide Action Network North America. Permission is granted to reproduce portions of this report, provided the title and publishing organizations—Pesticide Action Network and Californians for Pesticide Reform—are acknowledged. Our sincerest thanks to the Hesperian Foundation for providing many of the images used in these materials. Copyright © 2003 by the Hesperian Foundation. The Hesperian Foundation encourages others to copy, reproduce, or adapt to meet local needs any or all of this pamphlet provided that what is reproduced is distributed free or at cost—not for profit. -

Multiresidue Analysis of Pesticides in Avocado with Agilent Bond Elut EMR—Lipid by GC/MS/MS
Multiresidue Analysis of Pesticides in Avocado with Agilent Bond Elut EMR—Lipid by GC/MS/MS Application Note Food Testing and Agriculture Authors Abstract Limian Zhao and Derick Lucas Agilent Bond Elut Enhanced Matrix Removal—Lipid (EMR—Lipid) is the next Agilent Technologies, Inc. generation of sample preparation products, and is used in convenient, dispersive solid phase extraction (dSPE) for highly selective matrix removal without impacting analyte recovery, especially for high-fat samples. This study demonstrates the application of this novel product for the analysis of 23 GC-amenable pesticides in avocado by GC/MS/MS. The procedure involves a QuEChERS AOAC extraction followed by EMR—Lipid dSPE and polish salts. EMR—Lipid provides far superior matrix removal by weight, GC/MS full scan, and matrix effect determination when compared to C18/PSA and zirconia-based sorbents. Furthermore, less matrix is introduced into the analytical flow path. The data also demonstrate dramatically improved reproducibility for the analytes over 100 injections relative to C18/PSA and especially zirconia, which experience significant response deviations. EMR—Lipid is highly selective for lipids and does not negatively affect analyte recovery. Analyte recoveries are high and precision is outstanding. This work demonstrates that EMR—Lipid dSPE fits into a QuEChERS workflow and delivers fast, robust, and effective sample preparation with the most complete matrix removal available for multiresidue analysis of pesticides in avocado. Introduction removal sorbents. However, it does not target all lipid classes Pesticide residue analysis in food commodities is routine for and can retain analytes of interest [6,7]. Samples high in lipid many laboratories that use the Quick, Easy, Cheap, Effective, content may also require cleanup using solid phase extraction Rugged, and Safe (QuEChERS) method [1,2]. -
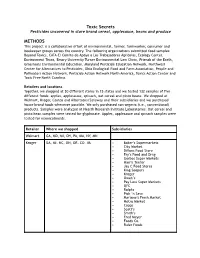
Food Testing Methods for Website
Toxic Secret s Pesticides uncovered in store brand cereal, applesauce, beans and produce METHODS This project is a collaborative effort of environmental, farmer, farmworker, consumer and beekeeper groups across the country. The following organizations submitted food samples: Beyond Toxics, CATA-El Comite de Apoyo a Los Trabajadores Agricolas, Ecology Center, Environment Texas, Emory University Turner Environmental Law Clinic, Friends of the Earth, Grassroots Environmental Education, Maryland Pesticide Education Network, Northwest Center for Alternatives to Pesticides, Ohio Ecological Food and Farm Association, People and Pollinators Action Network, Pesticide Action Network North America, Toxics Action Center and Toxic Free North Carolina. Retailers and locations Together, we shopped at 30 different stores in 15 states and we tested 132 samples of five different foods: apples, applesauce, spinach, oat cereal and pinto beans. We shopped at Walmart, Kroger, Costco and Albertsons/Safeway and their subsidiaries and we purchased house brand foods whenever possible. We only purchased non-organic (i.e., conventional) products. Samples were analyzed at Health Research Institute Laboratories. Oat cereal and pinto bean samples were tested for glyphosate. Apples, applesauce and spinach samples were tested for neonicotinoids. Retailer Where we shopped Subsidiaries Walmart GA, MD, MI, OH, PA, MA, NY, MN Kroger GA, MI, NC, OH, OR, CO, VA • Baker’s Supermarkets • City Market • Dillons Food Store • Fry’s Food and Drug • Gerbes Super Markets • Harris Teeter -

Pesticides and Honey Bees: State of the Science
Pesticides and Honey Bees: State of the Science MAY 2012 PESTICIDE ACTION NETWORK NORTH AMERICA Pes7cide Ac7on Network North America Pesticide Action Network North America (PAN North America) works to replace the use of hazardous pesticides with ecologically sound and socially just alternatives. As one of ?ive PAN Regional Centers worldwide, we link local and international consumer, labor, health, environment and agriculture groups into an international citizens’ action network. This network challenges the global proliferation of pesticides, defends basic rights to health and environmental quality, and works to ensure the transition to a just and viable society. Written by Heather Pilatic, PhD with research assistance from PANNA interns Juli Chamberlain and Neva Jacobs. Colleagues at Beyond Pesticides provided important annotations. Dr. Susan Kegley, consulting scientist for PANNA, and Dr. James Frazier, professor of entomology at Pennsylvania State served as helpful readers of early drafts. PANNA colleagues Kristin Schafer and Sara Knight contributed careful copyediting. Errors belong to the main author alone. Support for this work provided by the Ceres Foundation and PANNA supporting members. 49 Powell Street, Suite 500 San francisco, CA 94102 Tel: 415-981-1771 :: Fax: 415-981-1991 Pesticide Action Network www.panna.org :: www.pescideinfo.org Table of Contents Introduc*on ....................................................................................................................1 Overview :: Colony Collapse Disorder in context -

(PAN Asia Pacific): Highly Hazardous Pesticide Use and Impacts In
SAICM/OEWG.3/INF/39 Distr.: General 1 April 2019 Original: English Open-ended Working Group of the International Conference on Chemicals Management Third meeting Montevideo, 2–4 April 2019 Item 4(c) of the provisional agenda* Progress towards the achievement of the 2020 overall objectives of the sound management of chemicals: Emerging policy issues and other of concern Submission from the Pesticide Action Network Asia Pacific (PAN Asia Pacific): Highly Hazardous Pesticide Use and Impacts in Asia: The Need for Legally Binding Protocols Beyond 2020 Note by the secretariat The secretariat has the honour to circulate, in the annex to the present note, a report received from the Pesticide Action Network Asia Pacific (PAN Asia Pacific), on the highly hazardous pesticide use and impacts in Asia: the need for legally binding protocols beyond 2020. The report is presented in the annex as received from PAN Asia Pacific and has not been edited by the secretariat. * SAICM/OEWG.3/1. Highly Hazardous Pesticide Use and Impacts in Asia: The Need for Legally Binding Protocols Beyond 2020 A survey of 2025 small-scale farmers and agricultural workers was carried out in 7 Asian countries: Bangladesh, India, Indonesia, Malaysia, Pakistan, Philippines, Vietnam Poisoning & Use of Highly Hazardous Pesticides (HHPs) 7 out of every 10 farmers were suffering from symptoms of acute pesticide poisoning. 50 HHPs were in use, including benomyl, butachlor, carbendazim, carbofuran, carbosulfan, chlorfluazuron, chlorthalonil, chlorpyrifos, diazinon, dichlorvos, lambda-cyhalothrin, malathion, monocrotophos, paraquat, trichlorfon. Conditions of Use Users often lacked information on the pesticides they used; retailers sold pesticides in unlabeled plastic bags; or the plantation management provided workers pesticides without the labels. -

Chemical Trespass Pesticides in Our Bodies and Xcorporate Accountability
Chemical Trespass Pesticides in Our Bodies and XCorporate Accountability Kristin S. Schafer, Margaret Reeves, Skip Spitzer, Susan E. Kegley Pesticide Action Network North America May 2004 Chemical Trespass Pesticides in Our Bodies and Corporate Accountability Pesticide Action Network North America May 2004 Authors Kristin S. Schafer, Program Coordinator, Persistent Pesticides Margaret Reeves, PhD, Senior Scientist Skip Spitzer, Program Coordinator, Corporate Accountability Susan E. Kegley, PhD, Senior Scientist Acknowledgements This report reflects the efforts and expertise of many individuals both within Pesticide Action Network North America and among our partner organizations and institutions. Jessica Koll- meyer worked closely with the authors in researching and drafting the report, and Stephan Orme provided expertise on accessing and managing data. Monica Moore, Tanya Brown and Martha Olson Jarocki provided useful comments as the report was being developed and finalized, and Bob Sholtz helped strengthen our analytical approach to data analysis. Previous work of our colleague Marion Moses, M.D. cataloguing evidence of specific health effects of pesticides was invaluable in developing the health impacts discussion in this report. We also gratefully acknowledge the insights and suggestions provided by the reviewers of the report: Carol Dansereau, Michael DiBartolomeis, Joe DiGangi, Kim Hooper, Phil Howard, Sharyle Patton and Erika Schreder. Participation in the report’s review does not necessarily indicate endorsement by the reviewers or their organizations of the analyses, conclusions or recommendations presented. Thanks also go to Brenda J. Willoughby who formatted the report for publication, Mateo Rutherford and Roy Rojas of Berkeley Interpretation, Translation and Transcription Services (BITTS) who translated the Executive Summary into Spanish, and Cori Fay Traub, Martha Olson Jarocki, Travis Coan, Jessica Kollmeyer and Alexandra Latta for proofing and copy edit- ing.