Science Manuscript Template
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
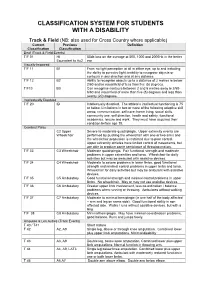
Disability Classification System
CLASSIFICATION SYSTEM FOR STUDENTS WITH A DISABILITY Track & Field (NB: also used for Cross Country where applicable) Current Previous Definition Classification Classification Deaf (Track & Field Events) T/F 01 HI 55db loss on the average at 500, 1000 and 2000Hz in the better Equivalent to Au2 ear Visually Impaired T/F 11 B1 From no light perception at all in either eye, up to and including the ability to perceive light; inability to recognise objects or contours in any direction and at any distance. T/F 12 B2 Ability to recognise objects up to a distance of 2 metres ie below 2/60 and/or visual field of less than five (5) degrees. T/F13 B3 Can recognise contours between 2 and 6 metres away ie 2/60- 6/60 and visual field of more than five (5) degrees and less than twenty (20) degrees. Intellectually Disabled T/F 20 ID Intellectually disabled. The athlete’s intellectual functioning is 75 or below. Limitations in two or more of the following adaptive skill areas; communication, self-care; home living, social skills, community use, self direction, health and safety, functional academics, leisure and work. They must have acquired their condition before age 18. Cerebral Palsy C2 Upper Severe to moderate quadriplegia. Upper extremity events are Wheelchair performed by pushing the wheelchair with one or two arms and the wheelchair propulsion is restricted due to poor control. Upper extremity athletes have limited control of movements, but are able to produce some semblance of throwing motion. T/F 33 C3 Wheelchair Moderate quadriplegia. Fair functional strength and moderate problems in upper extremities and torso. -

AFM&EPF2 Application with Exhibits Through 7.07
American Federation of Musicians and Employers Pension Fund Exhibit 7.07 Group 1 – 596 Custom CBAs 79801001 CHICAGO MASTER SINGERS DIVINE WORD CHAPEL CBA 2. Pension: THE EMPLOYER shall pay to the AMERICAN FEDERATION OF MUSICIANS AND EMPLOYERS' PENSION FUND ao amount equal to eleven ix,rcent (11%) of the Employe(s.gross payroll for all employees covered by this Agreement. Such payment shall be foiwarded to the Office of the Union during the week iol\owlng the week for which the payment is made. The Employer shall file contemporaneously with the aforesaid payment infonnation relating to the employees on whos~ behalf contributions are paid, including the amployee's name, so.cial security number, wages and such other lnformation which the Trustees of \he Fund may reasonably require. The Employer adopts and agrees to be bound by all the terms and aondluons of the Trust Agreement creating the AMERICAN FEDERATION OF MUSICIANS' AND EMPLOYERS' PENSION FUND, dated October 2, 1959, as amended fr-Om time to time, as fully as if the Employer were an ori9inal party thereto. The Employer hereby ratifies and agrees to be bound by an actions taken and to be taken by the Soard o!Trustees. pursuant to the Powers granted them by the Trust Agreement. The Fund shall provide pension benefits according to the AMERJCAN FEDERATION OF MUSICIANS' AND EMPLOYERS' PENSION PLAN, as amended by resofutlon dated December 3, 1964, and April 3, 1967.which said Amended Pension Plan Is attached hereto and made part hereof. In the event the Pension Plan shall be further amended, either In whole or in part, during the term of this Agreemen~ the revised Pension Plan shall be deemed to the Incorporation herein as if a part hereof. -

Framework for In-Field Analyses of Performance and Sub-Technique Selection in Standing Para Cross-Country Skiers
sensors Article Framework for In-Field Analyses of Performance and Sub-Technique Selection in Standing Para Cross-Country Skiers Camilla H. Carlsen 1,*, Julia Kathrin Baumgart 1, Jan Kocbach 1,2, Pål Haugnes 1 , Evy M. B. Paulussen 1,3 and Øyvind Sandbakk 1 1 Centre for Elite Sports Research, Department of Neuromedicine and Movement Science, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, 7491 Trondheim, Norway; [email protected] (J.K.B.); [email protected] (J.K.); [email protected] (P.H.); [email protected] (E.M.B.P.); [email protected] (Ø.S.) 2 NORCE Norwegian Research Centre AS, 5008 Bergen, Norway 3 Faculty of Health, Medicine & Life Sciences, Maastricht University, 6200 MD Maastricht, The Netherlands * Correspondence: [email protected]; Tel.: +47-452-40-788 Abstract: Our aims were to evaluate the feasibility of a framework based on micro-sensor technology for in-field analyses of performance and sub-technique selection in Para cross-country (XC) skiing by using it to compare these parameters between elite standing Para (two men; one woman) and able- bodied (AB) (three men; four women) XC skiers during a classical skiing race. The data from a global navigation satellite system and inertial measurement unit were integrated to compare time loss and selected sub-techniques as a function of speed. Compared to male/female AB skiers, male/female Para skiers displayed 19/14% slower average speed with the largest time loss (65 ± 36/35 ± 6 s/lap) Citation: Carlsen, C.H.; Kathrin found in uphill terrain. -

Before the Public Service Commission of the State of Missouri
Exhibit No.: Issues: Depreciation Witness: Brian C. Andrews Type of Exhibit: Direct Testimony Sponsoring Party: Missouri Industrial Energy Consumers Case No.: ER-2019-0335 Date Testimony Prepared: December 4, 2019 FILED March 19, 2020 Data Center BEFORE THE PUBLIC SERVICE COMMISSION Missouri Public OF THE STATE OF MISSOURI Service Commission ) In the Matter of Union Electric Company ) d/b/a Ameren Missouri's Tariffs to Decrease ) Case No. ER-2019-0335 Its Revenues for Electric Service. ) ------------) Direct Testimony and Schedules of Brian C. Andrews On behalf of Missouri Industrial Energy Consumers December 4, 2019 BRUIIAKr R & ASSOCIATES. INC. Project 10842 BEFORE THE PUBLIC SERVICE COMMISSION OF THE STATE OF MISSOURI ) In the Matter of Union Electric Company ) d/b/a Ameren Missouri's Tariffs to Decrease ) Case No. ER-2019-0335 Its Revenues for Electric Service. ) ) STATE OF MISSOURI ) ) ss COUNTY OF ST. LOUIS ) Affidavit of Brian C. Andrews Brian C. Andrews, being first duly sworn, on his oath states: 1. My name is Brian C. Andrews. I am a consultant with Brubaker & Associates, Inc., having its principal place of business at 16690 Swingley Ridge Road, Suite 140, Chesterfield, Missouri 63017. We have been retained by the Missouri Industrial Energy Consumers in this proceeding on their behalf. 2. Attached hereto and made a part hereof for all purposes are my direct testimony and schedules which were prepared in written form for introduction into evidence in Missouri Public Service Commission Case No. ER-2019-0335. 3. I hereby swear and affirm that the testimony and schedules are true and correct and that they show the matters and things that they purport to show. -

National Classification? 13
NATIONAL CL ASSIFICATION INFORMATION FOR MULTI CLASS SWIMMERS Version 1.2 2019 PRINCIPAL PARTNER MAJOR PARTNERS CLASSIFICATION PARTNERS Version 1.2 2019 National Swimming Classification Information for Multi Class Swimmers 1 CONTENTS TERMINOLOGY 3 WHAT IS CLASSIFICATION? 4 WHAT IS THE CLASSIFICATION PATHWAY? 4 WHAT ARE THE ELIGIBLE IMPAIRMENTS? 5 CLASSIFICATION SYSTEMS 6 CLASSIFICATION SYSTEM PARTNERS 6 WHAT IS A SPORT CLASS? 7 HOW IS A SPORT CLASS ALLOCATED TO AN ATHLETE? 7 WHAT ARE THE SPORT CLASSES IN MULTI CLASS SWIMMING? 8 SPORT CLASS STATUS 11 CODES OF EXCEPTION 12 HOW DO I CHECK MY NATIONAL CLASSIFICATION? 13 HOW DO I GET A NATIONAL CLASSIFICATION? 13 MORE INFORMATION 14 CONTACT INFORMATION 16 Version 1.2 2019 National Swimming Classification Information for Multi Class Swimmers 2 TERMINOLOGY Assessment Specific clinical procedure conducted during athlete evaluation processes ATG Australian Transplant Games SIA Sport Inclusion Australia BME Benchmark Event CISD The International Committee of Sports for the Deaf Classification Refers to the system of grouping athletes based on impact of impairment Classification Organisations with a responsibility for administering the swimming classification systems in System Partners Australia Deaflympian Representative at Deaflympic Games DPE Daily Performance Environment DSA Deaf Sports Australia Eligibility Criteria Requirements under which athletes are evaluated for a Sport Class Evaluation Process of determining if an athlete meets eligibility criteria for a Sport Class HI Hearing Impairment ICDS International Committee of Sports for the Deaf II Intellectual Impairment Inas International Federation for Sport for Para-athletes with an Intellectual Disability General term that refers to strategic initiatives that address engagement of targeted population Inclusion groups that typically face disadvantage, including people with disability. -

United States Olympic Committee and U.S. Department of Veterans Affairs
SELECTION STANDARDS United States Olympic Committee and U.S. Department of Veterans Affairs Veteran Monthly Assistance Allowance Program The U.S. Olympic Committee supports Paralympic-eligible military veterans in their efforts to represent the USA at the Paralympic Games and other international sport competitions. Veterans who demonstrate exceptional sport skills and the commitment necessary to pursue elite-level competition are given guidance on securing the training, support, and coaching needed to qualify for Team USA and achieve their Paralympic dreams. Through a partnership between the United States Department of Veterans Affairs and the USOC, the VA National Veterans Sports Programs & Special Events Office provides a monthly assistance allowance for disabled Veterans of the Armed Forces training in a Paralympic sport, as authorized by 38 U.S.C. § 322(d) and section 703 of the Veterans’ Benefits Improvement Act of 2008. Through the program the VA will pay a monthly allowance to a Veteran with a service-connected or non-service-connected disability if the Veteran meets the minimum VA Monthly Assistance Allowance (VMAA) Standard in his/her respective sport and sport class at a recognized competition. Athletes must have established training and competition plans and are responsible for turning in monthly and/or quarterly forms and reports in order to continue receiving the monthly assistance allowance. Additionally, an athlete must be U.S. citizen OR permanent resident to be eligible. Lastly, in order to be eligible for the VMAA athletes must undergo either national or international classification evaluation (and be found Paralympic sport eligible) within six months of being placed on the allowance pay list. -
![Downloaded As 2Ibz.Pdb [21] Protein from Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB)](https://docslib.b-cdn.net/cover/3309/downloaded-as-2ibz-pdb-21-protein-from-research-collaboratory-for-structural-bioinformatics-rcsb-protein-data-bank-pdb-1503309.webp)
Downloaded As 2Ibz.Pdb [21] Protein from Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB)
molecules Article Search for Novel Lead Inhibitors of Yeast Cytochrome bc1, from Drugbank and COCONUT Ozren Jovi´c* and Tomislav Šmuc Ruder¯ Boškovi´cInstitute, BijeniˇckaCesta 54, 10 000 Zagreb, Croatia; [email protected] * Correspondence: [email protected]; Tel.: +385-1-4561-085 Abstract: In this work we introduce a novel filtering and molecular modeling pipeline based on a fingerprint and descriptor similarity procedure, coupled with molecular docking and molecular dynamics (MD), to select potential novel quoinone outside inhibitors (QoI) of cytochrome bc1 with the aim of determining the same or different chromophores to usual. The study was carried out using the yeast cytochrome bc1 complex with its docked ligand (stigmatellin), using all the fungicides from FRAC code C3 mode of action, 8617 Drugbank compounds and 401,624 COCONUT compounds. The introduced drug repurposing pipeline consists of compound similarity with C3 fungicides and molecular docking (MD) simulations with final QM/MM binding energy determination, while aiming for potential novel chromophores and perserving at least an amide (R1HN(C=O)R2) or ester functional group of almost all up to date C3 fungicides. 3D descriptors used for a similarity test were based on the 280 most stable Padel descriptors. Hit compounds that passed fingerprint and 3D descriptor similarity condition and had either an amide or an ester group were submitted to docking where they further had to satisfy both Chemscore fitness and specific conformation constraints. This rigorous selection resulted in a very limited number of candidates that were forwarded to MD simulations and QM/MM binding affinity estimations by the ORCA DFT program. -

And Nitrogen-Codoped Porous Graphene
Journal of Catalysis 359 (2018) 242–250 Contents lists available at ScienceDirect Journal of Catalysis journal homepage: www.elsevier.com/locate/jcat Synergistic enhancement of oxygen reduction reaction with BC3 and graphitic-N in boron- and nitrogen-codoped porous graphene ⇑ ⇑ Li Qin a,b,d, Liancheng Wang a,b, , Xi Yang a,b, Ruimin Ding a,b, Zhanfeng Zheng a, Xiaohua Chen c, , ⇑ Baoliang Lv a,b, a State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China b CAS Key Laboratory of Carbon Materials, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China c School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400030, China d University of Chinese Academy of Sciences, Beijing 100049, China article info abstract Article history: Rational design and optimization of metal-free electrocatalysts for the oxygen reduction reaction (ORR) is Received 27 September 2017 crucial for fuel cells and metal-air batteries. However, identifying design principle that links the active Revised 4 January 2018 sites and their synergistic effects is far from satisfactory, especially for B,N-codoped graphene. Herein, Accepted 15 January 2018 we provide four B,N-codoped porous graphenes with tunable contents of pyridinic N, graphitic N, BC3 and C-B(N)O. BC3 shows multiple-fold specific activity compared with graphitic N and pyridinic N, while C-B(N)O offers no positive contribution. Density functional theory calculations indicate that the synergis- Keywords: tic effect between graphitic N and BC can effectively facilitate the reduction of O . These pinpoint that B, N-codoped porous graphene 3 2 graphitic N and BC are the main active sites among various nitrogen or/and boron doping configurations. -
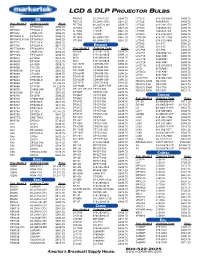
Lcd & Dlp Projector Bulbs
LCD & DLP PROJECTOR BULBS PB8263 5J.J2H01.001 $548.75 LV7320 610-289-6939 $498.75 3M PE5120 59.J9901.CG1 $561.25 LV7325 4638AA001 $498.75 Proj. Model # Ordering Code Price PE7765 60.J00720.001 $598.75 LV7345 610-293-2751 $498.75 540 EP7640ILK $486.25 SL703S 120VIP- $561.25 LV7350 7463A001AA $498.75 MP7630 EP7630LK $673.75 SL705S 120VIP- $561.25 LV7355 7463A001AA $498.75 MP7640 LAMP-029 $486.25 SL705X 120VIP- $561.25 LV7500 610-276-3010 $536.25 MP7640ILK EP7640ILK $486.25 VP150X 60.J0804.CB2 $561.25 LV7525E 610-282-2755 $498.75 MP7640ILK-S40 EP7640ILK $486.25 VT150X 60.J0804.CB2 $561.25 LV7545 610-293-5868 $536.25 MP7720 EP7720LK $836.25 LV7555 LV-LP17 $586.25 OXLIGHT MP7730 EP7630LK $673.75 B LV7565 LV-LP22 $623.75 Proj. Model # Ordering Code Price MP7730BAV EP7630BLK $723.75 LV-LP06 LV-LP06 $436.25 3600(A) 610-264-1943 $398.75 MP7740 LAMP-029 $486.25 LV-LP11 7463A001AA $498.75 3650 610-265-8828 $498.75 MP7740I EP7740ILK $498.75 LV-LP15 8441A001 $498.75 6000 610-265-8828 $498.75 MP8610 EP1830 $223.75 LV-LP18 9268A001 $498.75 6001 610-265-8828 $498.75 MP8620 EP1830 $223.75 LV-LP20 9431A001 $398.75 CD-450M CD450M-930 $586.25 MP8625 EP1890 $698.75 LV-S1-XL 610-293-8210 $498.75 CD-454 CP455M-930 $536.25 MP8630 CPL750 $461.25 LV-S2 8276A001 $498.75 CD-455M CP455M-930 $536.25 MP8635(B) EP-1625 $561.25 LV-S3(U) 9431A001 $398.75 CD-550M CD450M-930 $586.25 MP8640 CPL850 $498.75 LV-X2 8441A001 $498.75 CD-552M CP455M-930 $536.25 MP8647 EP8746LK $611.25 LV-X2THX 610-300-7267 $498.75 CD-555M CD450M-930 $586.25 MP8649 EP8749LK $511.25 REALIS SX50 RS-LP01 $623.75 CD-727X CD727X-930 $461.25 MP8650 EP1750 $1,223.75 REALIS SX6 RS-LP02 $623.75 CD-750M SP-LAMP-LP5F $561.25 MP8660 EP1750 $1,223.75 REALIS SX60 RS-LP03 $623.75 CP-13T, CP-33T CP13T-930 $498.75 MP8670 CP860-960 $523.75 REALIS X600 RS-LP02 $623.75 MP8725(B) EP-1625 $561.25 CP300T CP310T-930 $498.75 MP8740 EP2010 $586.25 CP305T CP13T-930 $498.75 CHRISTIE MP8745 CP860-960 $523.75 CP306T CP13T-930 $498.75 Proj. -

(VA) Veteran Monthly Assistance Allowance for Disabled Veterans
Revised May 23, 2019 U.S. Department of Veterans Affairs (VA) Veteran Monthly Assistance Allowance for Disabled Veterans Training in Paralympic and Olympic Sports Program (VMAA) In partnership with the United States Olympic Committee and other Olympic and Paralympic entities within the United States, VA supports eligible service and non-service-connected military Veterans in their efforts to represent the USA at the Paralympic Games, Olympic Games and other international sport competitions. The VA Office of National Veterans Sports Programs & Special Events provides a monthly assistance allowance for disabled Veterans training in Paralympic sports, as well as certain disabled Veterans selected for or competing with the national Olympic Team, as authorized by 38 U.S.C. 322(d) and Section 703 of the Veterans’ Benefits Improvement Act of 2008. Through the program, VA will pay a monthly allowance to a Veteran with either a service-connected or non-service-connected disability if the Veteran meets the minimum military standards or higher (i.e. Emerging Athlete or National Team) in his or her respective Paralympic sport at a recognized competition. In addition to making the VMAA standard, an athlete must also be nationally or internationally classified by his or her respective Paralympic sport federation as eligible for Paralympic competition. VA will also pay a monthly allowance to a Veteran with a service-connected disability rated 30 percent or greater by VA who is selected for a national Olympic Team for any month in which the Veteran is competing in any event sanctioned by the National Governing Bodies of the Olympic Sport in the United State, in accordance with P.L. -

The Paralympic Athlete Dedicated to the Memory of Trevor Williams Who Inspired the Editors in 1997 to Write This Book
This page intentionally left blank Handbook of Sports Medicine and Science The Paralympic Athlete Dedicated to the memory of Trevor Williams who inspired the editors in 1997 to write this book. Handbook of Sports Medicine and Science The Paralympic Athlete AN IOC MEDICAL COMMISSION PUBLICATION EDITED BY Yves C. Vanlandewijck PhD, PT Full professor at the Katholieke Universiteit Leuven Faculty of Kinesiology and Rehabilitation Sciences Department of Rehabilitation Sciences Leuven, Belgium Walter R. Thompson PhD Regents Professor Kinesiology and Health (College of Education) Nutrition (College of Health and Human Sciences) Georgia State University Atlanta, GA USA This edition fi rst published 2011 © 2011 International Olympic Committee Blackwell Publishing was acquired by John Wiley & Sons in February 2007. Blackwell’s publishing program has been merged with Wiley’s global Scientifi c, Technical and Medical business to form Wiley-Blackwell. Registered offi ce: John Wiley & Sons, Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK Editorial offi ces: 9600 Garsington Road, Oxford, OX4 2DQ, UK The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK 111 River Street, Hoboken, NJ 07030-5774, USA For details of our global editorial offi ces, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com/wiley-blackwell The right of the author to be identifi ed as the author of this work has been asserted in accordance with the UK Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. -

Distance of Antenna from Neighboring Houses (Feet): /UO' Distance of Antenna from Power Distribution Line Or Equipmmt
I' J Distance of antenna from neighboring houses (feet): /UO' Distance of antenna from power distribution line or equipmmt I I Made "---"- ------ .. ..*- . I I.. c .r qe * Page 1 of 2 +* - es Burtle ,: ,: Thursday, September 02,2004 258 PM James Burtle; Alan Scrim; Alan Stillwell; Bruce Franca; Karen Rackley iect Fw: BPL Interference complaint, new Ion-public: Fa M US^ OI@ *** Mginal Mesage- n: Doyle Hady [mailbo:dghardy@chw*net] i: Thursday, Septiember 02,2004 236 PM 4nh Wridt je.. BPL 1- le of complainant: Doyle.Hardy sign (if applicable): KSHTE ion location: 6521 Circleview Dr ,Ft Worth Tx, 76180 ling address (ifdifferen): 1, state, zip: Ft Worth TX, 76180 =phone: 817 -281-8670 Fm-d:[email protected] miption of Interference:- I was in contact with -K8NDS who at the time was opemting mobil station :ottonwood AZ. He was at Murphy’@ in Cottonwood. The interfence i hmthe local BPL jest made my signal unreadable. When the BPL was off I had a signal of S7 and very scription of station: My station is Kenwood TS-570D, running 100 ‘g of inwcrence: Dak Time Frequency Receive Interfering Description Mode signal 09/02/04 18OOGMT 14225 SSB 57 + was informedthat MHZ BPL totally made my signal unreadable. I I - :sage page 2 of 2 *' 8 I * es protect from harmful interference 's rules, Part 15 includes a definition of harmful interference. It can be found in S15.3 : "Harmful interference. Any emission, radiation or induction that endanger8 the functioning ). a radio navigation service or of other safety services or seriously degrades, obstructs, or eatedly interrupts a radiocommunication service operating in accordance with this chapter." < rules are very clear about the operation of Part 15 devices, too.