Exceptional Durable Response to Everolimus in a Patient with Biphenotypic Breast Cancer Harboring an STK11 Variant
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Inhibition of Mitochondrial Complex II in Neuronal Cells Triggers Unique
www.nature.com/scientificreports OPEN Inhibition of mitochondrial complex II in neuronal cells triggers unique pathways culminating in autophagy with implications for neurodegeneration Sathyanarayanan Ranganayaki1, Neema Jamshidi2, Mohamad Aiyaz3, Santhosh‑Kumar Rashmi4, Narayanappa Gayathri4, Pulleri Kandi Harsha5, Balasundaram Padmanabhan6 & Muchukunte Mukunda Srinivas Bharath7* Mitochondrial dysfunction and neurodegeneration underlie movement disorders such as Parkinson’s disease, Huntington’s disease and Manganism among others. As a corollary, inhibition of mitochondrial complex I (CI) and complex II (CII) by toxins 1‑methyl‑4‑phenylpyridinium (MPP+) and 3‑nitropropionic acid (3‑NPA) respectively, induced degenerative changes noted in such neurodegenerative diseases. We aimed to unravel the down‑stream pathways associated with CII inhibition and compared with CI inhibition and the Manganese (Mn) neurotoxicity. Genome‑wide transcriptomics of N27 neuronal cells exposed to 3‑NPA, compared with MPP+ and Mn revealed varied transcriptomic profle. Along with mitochondrial and synaptic pathways, Autophagy was the predominant pathway diferentially regulated in the 3‑NPA model with implications for neuronal survival. This pathway was unique to 3‑NPA, as substantiated by in silico modelling of the three toxins. Morphological and biochemical validation of autophagy markers in the cell model of 3‑NPA revealed incomplete autophagy mediated by mechanistic Target of Rapamycin Complex 2 (mTORC2) pathway. Interestingly, Brain Derived Neurotrophic Factor -

Protein ID Gene Symbol Unique + Razor Sequence Coverage
Dengjel et al. Mol. BioSyst., 2010, Supplementary Material (ESI) for Molecular BioSystems DOI: 10.1039/c003962d This journal is (c) The Royal Society of Chemistry, 2010 Supplementary Table 1: Fibroblast control. Cell lysates were prepared from unlabled (Arg0/Lys0) and labled (Arg10/Lys8 fibroblasts after 14 cell doublings in culture (D14). Shown are normalized log2 transformed protein ratios of identified protein identification a minimum of 2 peptides per protein has to be identified (unique + razor). The database used for identification decoy database based on IPI v. 3.59 containing in total 160,820 entries. As a criterium for identification the posterior errror probaility rate of identified proteins (PEP) is shown. The peptide numbers represent peptides used for quantitation. Unique + Razor Sequence Peptide Variability Protein ID Gene Symbol Coverage [%] PEP Log2 Ratio No. [%] Quantil IPI00021033; COL3A1 11.1 5.91E‐62 ‐4.651 4 165.2 Q1 IPI00761105; TADA2B 1.7 2.46E‐02 ‐4.494 2 57.9 Q1 IPI00152007 ARHGEF17 2.3 5.41E‐09 ‐3.687 2 92.5 Q1 IPI00300725;C KRT6A 27 6.82E‐61 ‐3.299 4 204.0 Q1 IPI00022229; APOB 0.7 5.68E‐29 ‐2.944 8 202.2 Q1 IPI00298860; LTF 51 8.02E‐197 ‐2.834 13 105.6 Q1 IPI00384444; KRT14 36.4 6.18E‐85 ‐2.542 13 143.3 Q1 IPI00305461; ITIH2 5 2.86E‐09 ‐1.174 3 367.3 Q1 IPI00420088; KIAA0174 7.5 9.34E‐03 ‐0.892 2 62.3 Q1 IPI00012887; CTSL1 12 3.13E‐21 ‐0.618 3 47.8 Q1 IPI00375714; HSPB7 17.1 1.31E‐05 ‐0.615 2 69.5 Q1 IPI00384140 ESR2 10.6 4.35E‐04 ‐0.551 3 8.6 Q1 IPI00300599; CTSK 46.5 <1.0E‐304 ‐0.515 9 11.7 Q1 IPI00167949; -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Protein-Protein Interactions Among Signaling Pathways May Become New Therapeutic Targets in Liver Cancer (Review)
ONCOLOGY REPORTS 35: 625-638, 2016 Protein-protein interactions among signaling pathways may become new therapeutic targets in liver cancer (Review) XIAO ZHANG1*, YULAN WANG1*, Jiayi WANG1,2 and FENYONG SUN1 1Department of Clinical Laboratory Medicine, Shanghai Tenth People's Hospital of Tongji University, Shanghai 200072; 2Translation Medicine of High Institute, Tongji University, Shanghai 200092, P.R. China Received May 29, 2015; Accepted July 6, 2015 DOI: 10.3892/or.2015.4464 Abstract. Numerous signaling pathways have been shown to be 1. Introduction dysregulated in liver cancer. In addition, some protein-protein interactions are prerequisite for the uncontrolled activation Liver cancer is the sixth most common cancer and the second or inhibition of these signaling pathways. For instance, in most common cause of cancer-associated mortality world- the PI3K/AKT signaling pathway, protein AKT binds with wide (1). Approximately 75% of all primary liver cancer types a number of proteins such as mTOR, FOXO1 and MDM2 to are hepatocellular carcinoma (HCC) that formed from liver play an oncogenic role in liver cancer. The aim of the present cells. Liver cancer can be formed from other structures in review was to focus on a series of important protein-protein the liver such as bile duct, blood vessels and immune cells. interactions that can serve as potential therapeutic targets Secondary liver cancer is a result of metastasis of cancer from in liver cancer among certain important pro-carcinogenic other body sites into the liver. The major cause of primary liver signaling pathways. The strategies of how to investigate and cancer is viral infection with either hepatitis C virus (HCV) analyze the protein-protein interactions are also included in or hepatitis B virus (HBV), which leads to massive inflamma- this review. -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior. -
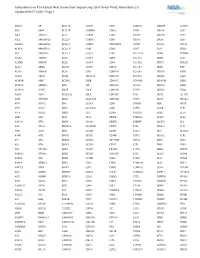
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1 ABCC3 AR BCL11A CANT1 CDK1 CMKLR1 DAB2IP DUSP9 ABI1 ARAF BCL11B CAPRIN1 CDK12 CNBP DACH1 E2F1 ABL1 ARFRP1 BCL2 CAPZB CDK2 CNOT2 DACH2 E2F3 ABL2 ARHGAP20 BCL2A1 CARD11 CDK4 CNTN1 DAXX EBF1 ABLIM1 ARHGAP26 BCL2L1 CARM1 CDK5RAP2 CNTRL DCLK2 ECT2L ACACA ARHGEF12 BCL2L11 CARS CDK6 COG5 DCN EDIL3 ACE ARHGEF7 BCL2L2 CASC5 CDK7 COL11A1 DDB1 EDNRB ACER1 ARID1A BCL3 CASP3 CDK8 COL1A1 DDB2 EED ACSBG1 ARID1B BCL6 CASP7 CDK9 COL1A2 DDIT3 EEFSEC ACSL3 ARID2 BCL7A CASP8 CDKL5 COL3A1 DDR2 EGF ACSL6 ARID5B BCL9 CAV1 CDKN1A COL6A3 DDX10 EGFR ACVR1 ARIH2 BCOR CBFA2T3 CDKN1B COL9A3 DDX20 EGR1 ACVR1B ARNT BCORL1 CBFB CDKN1C COMMD1 DDX39B EGR2 ACVR1C ARRDC4 BCR CBL CDKN2A COX6C DDX3X EGR3 ACVR2A ASMTL BDNF CBLB CDKN2B CPNE1 DDX41 EGR4 ADD3 ASPH BHLHE22 CBLC CDKN2C CPS1 DDX5 EIF1AX ADM ASPSCR1 BICC1 CCDC28A CDKN2D CPSF6 DDX6 EIF4A2 AFF1 ASTN2 BIN1 CCDC6 CDX1 CRADD DEK EIF4E AFF3 ASXL1 BIRC3 CCDC88C CDX2 CREB1 DGKB ELF3 AFF4 ASXL2 BIRC6 CCK CEBPA CREB3L1 DGKI ELF4 AGR3 ATF1 BLM CCL2 CEBPB CREB3L2 DGKZ ELK4 AHCYL1 ATF3 BMP4 CCNA2 CEBPD CREBBP DICER1 ELL AHI1 ATG13 BMPR1A CCNB1IP1 CEBPE CRKL DIRAS3 ELN AHR ATG5 BRAF CCNB3 CENPF CRLF2 DIS3 ELOVL2 AHRR ATIC BRCA1 CCND1 CENPU CRTC1 DIS3L2 ELP2 AIP ATL1 BRCA2 CCND2 CEP170B CRTC3 DKK1 EML1 AK2 ATM BRCC3 CCND3 CEP57 CSF1 DKK2 EML4 AK5 ATP1B4 BRD1 CCNE1 CEP85L CSF1R DKK4 ENPP2 AKAP12 ATP8A2 BRD3 CCNG1 CHCHD7 CSF3 DLEC1 EP300 AKAP6 ATR BRD4 CCT6B CHD2 CSF3R DLL1 EP400 AKAP9 ATRNL1 BRIP1 CD19 CHD4 CSNK1A1 DLL3 -

Inhibition of RPTOR Overcomes Resistance to EGFR Inhibition in Triple-Negative Breast Cancer Cells
828 INTERNATIONAL JOURNAL OF ONCOLOGY 52: 828-840, 2018 Inhibition of RPTOR overcomes resistance to EGFR inhibition in triple-negative breast cancer cells KYU SIC YOU1*, YONG WEON YI2*, SAHNG-JUNE KWAK3 and YEON-SUN SEONG1,3,4 1Graduate School of Convergence Medical Science, Dankook University, Cheonan 31116; 2ExoCoBio Inc, Seoul 08594; 3Department of Biochemistry, College of Medicine, Dankook University; 4Department of Nanobiomedical Science and BK21 PLUS Global Research Center for Regenerative Medicine, Dankook University, Cheonan 31116, Republic of Korea Received September 14, 2017; Accepted January 8, 2018 DOI: 10.3892/ijo.2018.4244 Abstract. Triple-negative breast cancer (TNBC) cells frequently protein S6 (RPS6), a marker of cell proliferation and target exhibit activated growth factor signaling and resistance to substrate of the AKT-mTOR signaling pathway. In addition, inhibitors for epidermal growth factor receptor (EGFR), despite gefitinib markedly reduced the viability of MDA-MD-231 and the overexpression of EGFR protein, and this is associated HS578T cells when regulatory-associated protein of mTOR with a malignant behavior and a poor prognosis. In this study, (RPTOR) was suppressed by siRNA-based knockdown (KD). to elucidate the underlying mechanisms of resistance to EGFR These results thus suggest that RPTOR mediates, at least inhibitor and identify inhibitors that exert a synergistic effect partially, the resistance to EGFR inhibition in TNBC cells. with EGFR inhibition, we examined the inhibitory effects of Therefore, targeting the mTOR complex 1 (mTORC1) pathway selected protein kinase inhibitors (PKIs) in combination with may be a potential strategy for the treatment of EGFR-resistant gefitinib on the viability of a mesenchymal stem-like (MSL) TNBC. -

Oncogenic Activation of PI3K-AKT-Mtor Signaling Suppresses Ferroptosis Via SREBP-Mediated Lipogenesis
Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis Junmei Yia,1, Jiajun Zhub,1, Jiao Wua,c, Craig B. Thompsonb,2, and Xuejun Jianga,2 aCell Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065; bCancer Biology and Genetics Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065; and cDepartment of Cell Biology, School of Basic Medicine, Air Force Medical University, 710032 Xi’an, China Edited by Xiaodong Wang, National Institute of Biological Sciences, Beijing, China, and approved October 12, 2020 (received for review August 13, 2020) Ferroptosis, a form of regulated necrosis driven by iron-dependent genetic backgrounds. However, the role that individual tumorigenic peroxidation of phospholipids, is regulated by cellular metabolism, mutations play in the regulation of the sensitivity of a given cancer redox homeostasis, and various signaling pathways related to can- to ferroptosis is just beginning to be investigated. cer. In this study, we found that activating mutation of phospha- Sterol regulatory element-binding protein 1 (SREBP1) is a tidylinositol 3-kinase (PI3K) or loss of phosphatase and tensin master regulator of lipid homeostasis and glucose metabolism homolog deleted on chromosome 10 (PTEN) function, highly fre- (20, 21). As a transendoplasmic reticular membrane protein, quent events in human cancer, confers ferroptosis resistance in SREBP1 senses various metabolic signals and is activated by cancer cells, and that inhibition of the PI3K-AKT-mTOR -

Supplemental Solier
Supplementary Figure 1. Importance of Exon numbers for transcript downregulation by CPT Numbers of down-regulated genes for four groups of comparable size genes, differing only by the number of exons. Supplementary Figure 2. CPT up-regulates the p53 signaling pathway genes A, List of the GO categories for the up-regulated genes in CPT-treated HCT116 cells (p<0.05). In bold: GO category also present for the genes that are up-regulated in CPT- treated MCF7 cells. B, List of the up-regulated genes in both CPT-treated HCT116 cells and CPT-treated MCF7 cells (CPT 4 h). C, RT-PCR showing the effect of CPT on JUN and H2AFJ transcripts. Control cells were exposed to DMSO. β2 microglobulin (β2) mRNA was used as control. Supplementary Figure 3. Down-regulation of RNA degradation-related genes after CPT treatment A, “RNA degradation” pathway from KEGG. The genes with “red stars” were down- regulated genes after CPT treatment. B, Affy Exon array data for the “CNOT” genes. The log2 difference for the “CNOT” genes expression depending on CPT treatment was normalized to the untreated controls. C, RT-PCR showing the effect of CPT on “CNOT” genes down-regulation. HCT116 cells were treated with CPT (10 µM, 20 h) and CNOT6L, CNOT2, CNOT4 and CNOT6 mRNA were analysed by RT-PCR. Control cells were exposed to DMSO. β2 microglobulin (β2) mRNA was used as control. D, CNOT6L down-regulation after CPT treatment. CNOT6L transcript was analysed by Q- PCR. Supplementary Figure 4. Down-regulation of ubiquitin-related genes after CPT treatment A, “Ubiquitin-mediated proteolysis” pathway from KEGG.