Mapping the Sex Determination Locus in the Hāpuku (Polyprion Oxygeneios) Using Ddrad Sequencing Jeremy K
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Appendices Appendices
APPENDICES APPENDICES APPENDIX 1 – PUBLICATIONS SCIENTIFIC PAPERS Aidoo EN, Ute Mueller U, Hyndes GA, and Ryan Braccini M. 2015. Is a global quantitative KL. 2016. The effects of measurement uncertainty assessment of shark populations warranted? on spatial characterisation of recreational fishing Fisheries, 40: 492–501. catch rates. Fisheries Research 181: 1–13. Braccini M. 2016. Experts have different Andrews KR, Williams AJ, Fernandez-Silva I, perceptions of the management and conservation Newman SJ, Copus JM, Wakefield CB, Randall JE, status of sharks. Annals of Marine Biology and and Bowen BW. 2016. Phylogeny of deepwater Research 3: 1012. snappers (Genus Etelis) reveals a cryptic species pair in the Indo-Pacific and Pleistocene invasion of Braccini M, Aires-da-Silva A, and Taylor I. 2016. the Atlantic. Molecular Phylogenetics and Incorporating movement in the modelling of shark Evolution 100: 361-371. and ray population dynamics: approaches and management implications. Reviews in Fish Biology Bellchambers LM, Gaughan D, Wise B, Jackson G, and Fisheries 26: 13–24. and Fletcher WJ. 2016. Adopting Marine Stewardship Council certification of Western Caputi N, de Lestang S, Reid C, Hesp A, and How J. Australian fisheries at a jurisdictional level: the 2015. Maximum economic yield of the western benefits and challenges. Fisheries Research 183: rock lobster fishery of Western Australia after 609-616. moving from effort to quota control. Marine Policy, 51: 452-464. Bellchambers LM, Fisher EA, Harry AV, and Travaille KL. 2016. Identifying potential risks for Charles A, Westlund L, Bartley DM, Fletcher WJ, Marine Stewardship Council assessment and Garcia S, Govan H, and Sanders J. -

Reproductive Biology of Southwestern Atlantic Wreckfish, Polyprion
Environmental Biology of Fishes 68: 163–173, 2003. © 2003 Kluwer Academic Publishers. Printed in the Netherlands. Reproductive biology of southwestern Atlantic wreckfish, Polyprion americanus (Teleostei: Polyprionidae) Monicaˆ B. Peresa & Sandro Klippelb aServic¸o da Regiao˜ Litoral, DQA, FEPAM Rua Carlos Chagas 55 sala 707, Centro Porto Alegre, RS 90.030-020, Brazil (e-mail: [email protected]) bTalha-mar Projetos Ambientais, Cons. d’Avila 190 Porto Alegre, RS 91040-450, Brazil Received 5 February 2003 Accepted 12 July 2003 Key words: sexual maturity, fecundity, spawning aggregation, spawning season, migration, deep-sea fish, life-cycle closure, southern Brazil Synopsis We report on the reproductive biology of southwestern Atlantic wreckfish. Females mature first at 77.9 cm total length (TL) (10.4 years) and all are mature by 90 cm TL (15.2 years). Males mature first at 74.9 cm (9 years) and all are mature by 80 cm TL (10.9 years). The wreckfish is a gonochoristic multiple spawner and the gonadal cycle is synchronized at the population level. Spawning occurs from late July to early October along the continental slope (>300 m). Ovarian fecundity varies from 3 to 11.9 million (135–311 oocytes × g−1) and increases exponentially with length. Spawning at western boundary current systems, maintained by homing of adults, is a basic requirement for self-sustaining populations of this species. Introduction are mature and juveniles may live more than 2 years at the sea surface before recruitment to the bottom The wreckfish, Polyprion americanus (Bloch and (Sedberry et al. 1999). Despite its worldwide distri- Schneider, 1801), is a large, long-lived demersal teleost bution and high commercial importance, there is no that inhabits continental and oceanic island slopes of information on reproductive cycle, fecundity, spawn- temperate and subtropical waters of the northern and ing sites and season of these fish. -

Kaikōura Marine Area If It Specified in the Regulations Has Not Had One-Third of the Telson Cut Off
KAIKŌURA Changes to the MARINE recreational AREA fishing rules Sperm whale © Whale Watch Kaikoura Sperm whale © Whale Watch To help keep fish stocks sustainable and protect against illegal fishing and poaching, the Kaikōura Marine Area has been introduced. Within the Marine Area (blue line on the map) there are Marine changes to the recreational Area fishing rules. What are the new rules? » The recreational daily bag limits for some finfish and shellfish species have been reduced. » The size limits for blue cod and sea perch have changed. » Daily limits for the harvest of Sanctuary bladder kelp and karengo have Marine Reserve been introduced. Mātaitai Taiāpure » A prohibition on the taking of red Marine Area moki. » The introduction of ‘telson clipping’ and an accumulation limit for spiny rock lobster. Turn over for a summary of the new rules. These are not the full rules – for a full version visit www.mpi.govt.nz Finfish Maximum daily limit Minimum length Species What is the ‘telson clipping’ rule? per fisher (cm) The telson is the central part of the tail fan on a Blue cod 6 33 rock lobster. Tarakihi 10 25 Telson clipping is a way of marking spiny rock Sea Perch 20 26 lobster to make it clear that they have been Kahawai 10 No limit recreationally caught. One-third of the telson is cut Butterfish 10 35 off so that it is noticeably shorter than the other Red Moki NO TAKE sections of the tail fan. (See the picture below). Blue Moki 10 40 Rig 3 No limit School shark 3 No limit Kingfish 75 Bass and Hapuku Daily combined bag No limit limit of five per person Bluenose No limit with a maximum of 3 Ling of any one species No limit Albacore tuna No limit Game sharks (seven-gilled shark, mako shark, blue shark, 1 game shark No limit hammerhead shark, porbeagle total One-third of the shark, thresher shark) central telson Shellfish has been clipped. -

Australia: Reconstructing Estimates of Total Fisheries Removal, 1950-2010
Fisheries Centre The University of British Columbia Working Paper Series Working Paper #2015 - 02 Australia: Reconstructing estimates of total fisheries removal, 1950-2010 Kristin Kleisner, Ciara Brennan, Anna Garland, Stephanie Lingard, Sean Tracey, Phil Sahlqvist, Angelo Tsolos, Daniel Pauly, and Dirk Zeller Year: 2015 Email: [email protected] ; [email protected] This working paper is made available by the Fisheries Centre, University of British Columbia, Vancouver, BC, V6T 1Z4, Canada. Australia - Kleisner et al. 1 AUSTRALIA: RECONSTRUCTING ESTIMATES OF TOTAL FISHERIES REMOVALS 1950-2010 Kristin Kleisnera, Ciara Brennana, Anna Garlandb, Stephanie Lingarda, Sean Traceyc, Phil Sahlqvistd, Angelo Tsolose, Daniel Paulya, and Dirk Zellera a Sea Around Us, Fisheries Centre, University of British Columbia, 2202 Main Mall, Vancouver, V6T 1Z4, Canada b Fisheries Queensland, Department of Agriculture, Fisheries and Forestry, GPO Box 46, Brisbane, Qld 4001, Australia c Institute for Marine and Antarctic Studies, University of Tasmania, Private bag 49, Hobart, Tasmania 7001, Australia d Fisheries and Risk Analysis Branch, ABARES, GPO Box 1563, Canberra ACT 2601, Australia e SARDI SA Aquatic Sciences Centre, Fisheries – Information Services, PO Box 120, Henley Beach, SA 5022 Australia [email protected]; kristen.kleisner @noaa.gov; c [email protected]; [email protected]; [email protected]; [email protected]; [email protected]; [email protected]; [email protected] ; [email protected] ABSTRACT Australia’s commercial fisheries are of significant value to the Australian economy, with the twenty Commonwealth fisheries alone worth around AUD$320 million in production value. -

Biology, Stock Status and Management Summaries for Selected Fish Species in South-Western Australia
Fisheries Research Report No. 242, 2013 Biology, stock status and management summaries for selected fish species in south-western Australia Claire B. Smallwood, S. Alex Hesp and Lynnath E. Beckley Fisheries Research Division Western Australian Fisheries and Marine Research Laboratories PO Box 20 NORTH BEACH, Western Australia 6920 Correct citation: Smallwood, C. B.; Hesp, S. A.; and Beckley, L. E. 2013. Biology, stock status and management summaries for selected fish species in south-western Australia. Fisheries Research Report No. 242. Department of Fisheries, Western Australia. 180pp. Disclaimer The views and opinions expressed in this publication are those of the authors and do not necessarily reflect those of the Department of Fisheries Western Australia. While reasonable efforts have been made to ensure that the contents of this publication are factually correct, the Department of Fisheries Western Australia does not accept responsibility for the accuracy or completeness of the contents, and shall not be liable for any loss or damage that may be occasioned directly or indirectly through the use of, or reliance on, the contents of this publication. Fish illustrations Illustrations © R. Swainston / www.anima.net.au We dedicate this guide to the memory of our friend and colleague, Ben Chuwen Department of Fisheries 3rd floor SGIO Atrium 168 – 170 St Georges Terrace PERTH WA 6000 Telephone: (08) 9482 7333 Facsimile: (08) 9482 7389 Website: www.fish.wa.gov.au ABN: 55 689 794 771 Published by Department of Fisheries, Perth, Western Australia. Fisheries Research Report No. 242, March 2013. ISSN: 1035 - 4549 ISBN: 978-1-921845-56-7 ii Fisheries Research Report No.242, 2013 Contents ACKNOWLEDGEMENTS ............................................................................................... -
Recreational Fishing Guide 2021
Department of Primary Industries and Regional Development Recreational fishing guide 2021 New rules apply from 1 July 2021 see page 3 for details Includes Statewide bag and size limits for Western Australia, and Recreational Fishing from Boat Licence information Published June 2021 Page i Important disclaimer The Director General of the Department of Primary Industries and Regional Development (DPIRD) and the State of Western Australia accept no liability whatsoever by reason of negligence or otherwise arising from the use or release of this information or any part of it. This publication is to provide assistance or information. It is only a guide and does not replace the Fish Resources Management Act 1994 or the Fish Resources Management Regulations 1995. It cannot be used as a defence in a court of law. The information provided is current at the date of printing but may be subject to change. For the most up-to-date information on fishing and full details of legislation contact select DPIRD offices or visit dpird.wa.gov.au Copyright © State of Western Australia (Department of Primary Industries and Regional Development) 2021 Front cover photo: Tourism WA Department of Primary Industries and Regional Development Gordon Stephenson House, 140 William Street, Perth WA 6000 +61 1300 374 731 [email protected] dpird.wa.gov.au Page ii Contents Fish for the future .............................................2 Using this guide .................................................2 Changes to the rules – 2021 .............................3 -

Hapuku (Polypm'onoxygeneios) and Bass (P.Americanus)
ISSN 1175-1584 MINISTRY OF FISHERIES re Tautiaki i nga tini a Tangama A description of the New Zealand fisheries for the two groper species, hapuku (Polypm'onoxygeneios) and bass (P.americanus) . L. J. Paul New Zealand Fisheries Assessment Report 2002113 March 2002 A description of the New Zealand fisheries for the two groper species, hapuku (Polyprion oxygenezbs) and bass (P. ameficanza) L. J. Paul NIWA PO Box 14 901 Wellington New Zealand Fisheries Assessment Report 2002113 March 2002 Published by Ministry of Fisheries Wellington 2002 ISSN 1175-1584 0 Ministry of Fisheries 2002 Citation: Paul, L.J. (2002). A description of the New Zealand fisheries for the two groper species, hapuku (Polyprion axygeneios) and bass (P. mnericanus). New Zealand Fi3heries Assessment Report 2002/13.47 p. This series continues the informal New Zealand Fisheries Assessment Research Document series which ceased at the end of 1999. EXECUTIVE SUMMARY Paul, L.J. (2002). A description of the New Zealand fisheries for the two groper species, hapukn (Polyprion oxygeneios) and bass (P. americnnus). New Zealand Fisheries Assessment Report 200WJ. 47 p. The groper fishery, based on two species (bass, hapuku), is relatively small. Landings reached 2500 t in the early 1980s, but were reduced to 1000 1500 t by the Quota Management System (QMS). Both species are valuable, and catches are made by many (500 700) fishers. Landings around the New Zealand mainland are close to and constrained by regional TACCs. This report describes the main features of the groper fshery. The preQMS fishery is briefly described, in terms of catch (= landing) by region and method. -

Kaikoura: Historical Background
Kaikoura: Historical Background Jim McAloon Lecturer, Human Sciences Division, Lincoln University. [email protected] David G Simmons Reader in Tourism, Human Sciences Division, Lincoln University. [email protected] John R Fairweather Senior Research Officer in the Agribusiness and Economics Research Unit, Lincoln University. [email protected] September 1998 ISSN 1174-670X Tourism Research and Education Centre (TREC) Report No. 1 Contents LIST OF FIGURES iii ACKNOWLEDGEMENTS iv SUMMARY v CHAPTER 1 KAIKOURA – HISTORICAL OVERVIEW.............................................. 1 1.1 Geography.......................................................................................... 1 1.2 Māori Settlement................................................................................ 1 1.3 Crown Purchases................................................................................ 4 1.4 Pastoralism......................................................................................... 6 1.5 Small Farming.................................................................................... 7 1.6 Conclusion: An Enduring Pattern ...................................................... 9 REFERENCES ...................................................................................................................... 13 ii List of Figures Figure No. Page No. 1. Areas of historical and Māori significance in the Kaikoura district..............2 iii Acknowledgements Funding for this research was provided by the Foundation for Research, -

Kosher Fish Around the World
KOSHER FISH AROUND THE WORLD Fresh and frozen kosher species of fish, either whole or filleted, are permitted provided that some skin (bearing easily detachable scales) is attached. All minced fish and products containing fish require Rabbinical supervision. The following varieties of fish are kosher. Here we list commonly available fish in 21 countries, translating the names where necessary. For lists of kosher fish in other countries, contact the KLBD on 020 8343 6247. EUROPEAN COUNTRIES UK CYPRUS CZECH REPUBLIC Anchovy Antzouva Ancovicka Bass Lavraki Okoun Bream Tsipoura Prazama Brill - - Carp - Kapr Cod - Treska Dab - Platejs Grayling - Lipan Haddock - Treska Halibut Bacceliaos Platyz Herring - Sled Mackerel - Makrela Mullet Scoumbri - Mullet Red - Parmice Perch Cephalos Okoun Pike Barbouni Stika Plaice - Playtz Pollack Sardella - Roach - Belicka Salmon - Losos Sardine - Sardinka Sole - Plotice Sprat - Sprota Tench Glossa Lin Trout - Pstruh Tuna Tonos Tunak Whiting - Merlan UK DENMARK FRANCE Anchovy Ansjos Anchois Bass Bars Bar Commun Bream Brasen Breme Brill Slethvarre Barbue Carp Karpe Carpe Cod Torsk Cabillaud Dab Ising Limande Flounder Skrubbe Flet Grayling - Ombre Gurnard Knurhane Grondin Haddock Kuller Aiglefin Hake Kulmule Merlu Halibut Helleyflynder Fletan Herring Sild Hareng Ling Lange Lingue Mackerel Makrel Maquereau Mullet Multe Mulet Perch Aborre Perche Pike Gedde Brochet Pilchard Sardin Pilchard Plaice Rodspaette Plie, Carrelet Pollack Lubbe Lieu Jaune Red Snapper - Rouget Roach Skalle Gardon Salmon Laks Saumon Sardine -
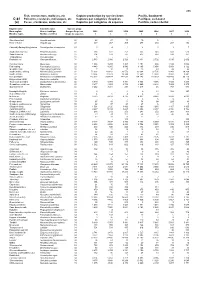
Fish, Crustaceans, Molluscs, Etc Capture Production by Species
495 Fish, crustaceans, molluscs, etc Capture production by species items Pacific, Southwest C-81 Poissons, crustacés, mollusques, etc Captures par catégories d'espèces Pacifique, sud-ouest (a) Peces, crustáceos, moluscos, etc Capturas por categorías de especies Pacífico, sudoccidental English name Scientific name Species group Nom anglais Nom scientifique Groupe d'espèces 2002 2003 2004 2005 2006 2007 2008 Nombre inglés Nombre científico Grupo de especies t t t t t t t Short-finned eel Anguilla australis 22 28 27 13 10 5 ... ... River eels nei Anguilla spp 22 337 267 209 277 210 207 152 Chinook(=Spring=King)salmon Oncorhynchus tshawytscha 23 0 4 1 2 1 1 7 Southern lemon sole Pelotretis flavilatus 31 238 322 251 335 348 608 513 Sand flounders Rhombosolea spp 31 204 193 187 437 514 530 351 Tonguefishes Cynoglossidae 31 3 - - - - - - Flatfishes nei Pleuronectiformes 31 2 580 2 986 2 729 3 431 2 702 3 015 2 602 Common mora Mora moro 32 1 308 1 234 1 403 1 154 986 1 180 1 088 Red codling Pseudophycis bachus 32 4 443 8 265 9 540 8 165 5 854 5 854 6 122 Grenadier cod Tripterophycis gilchristi 32 7 10 13 13 43 29 26 Southern blue whiting Micromesistius australis 32 72 203 43 812 26 576 30 304 32 735 23 943 29 268 Southern hake Merluccius australis 32 13 834 22 623 19 344 12 560 12 858 13 892 8 881 Blue grenadier Macruronus novaezelandiae 32 215 302 209 414 147 032 134 145 119 329 103 489 96 119 Ridge scaled rattail Macrourus carinatus 32 - - - - - 9 14 Thorntooth grenadier Lepidorhynchus denticulatus 32 5 349 5 304 6 341 3 855 4 056 3 725 3 264 Grenadiers, rattails nei Macrouridae 32 3 877 4 253 3 732 2 660 2 848 7 939 8 970 Gadiformes nei Gadiformes 32 3 252 3 281 298 1 217 46 767 886 Broadgilled hagfish Eptatretus cirrhatus 33 2 - 0 0 11 508 347 Sea catfishes nei Ariidae 33 4 6 4 4 4 .. -

Recreational Fishing Identification Guide
Department of Primary Industries and Regional Development Recreational fishing identification guide June 2020 Contents About this guide.................................................................................................. 1 Offshore demersal .............................................................................................. 3 Inshore demersal ................................................................................................ 4 Nearshore .........................................................................................................12 Estuarine ..........................................................................................................19 Pelagic ..............................................................................................................20 Sharks ..............................................................................................................23 Crustaceans .....................................................................................................25 Molluscs............................................................................................................27 Freshwater........................................................................................................28 Cover: West Australian dhufish Glaucosoma hebraicum. Photo: Mervi Kangas. Published by Department of Primary Industries and Regional Development, Perth, Western Australia. Fisheries Occasional Publication No. 103, sixth edition, June 2020. ISSN: 1447 – 2058 (Print) -

02 Francis FISH 97(2)
227 Abstract.–Polyprion oxygeneios (fam- Age and growth estimates for ily Polyprionidae) is fished commer- cially and recreationally in the south- New Zealand hapuku, Polyprion oxygeneios ern Indian and Pacific Oceans. Esti- mates of growth rate, age at maturity and recruitment, and longevity are re- Malcolm P. Francis quired for fishery management. We Kevin P. Mulligan used thin otolith sections to age P. oxygeneios from New Zealand, where it Nick M. Davies is known as hapuku. Growth bands were difficult to count, leading to low Michael P. Beentjes counting precision and, for some age National Institute of Water and Atmospheric Research groups, to a small between- and within- P. O. Box 14–901, Wellington, New Zealand reader ageing bias. These problems, E-mail address (for M. P. Francis): [email protected] however, had little effect on the shape of growth curves fitted to length-at-age data. An oxytetracycline injection ex- periment supported our hypothesis of annual deposition of an opaque-hyaline band pair, but further validation is re- Polyprion oxygeneios (Schneider, growth of P. americanus from the quired. Independently derived von 1801) (family Polyprionidae) is a Bertalanffy growth curves (from length- north-west Atlantic is currently frequency data) and growth-rate esti- large demersal fish that inhabits underway (Sedberry et al., 1994, mates (from tag-recapture data) for temperate and subtropical waters of 1996; Manooch and Potts1). How- young hapuku agreed well with esti- the southern Indian and Pacific ever, otolith bands have proven dif- mates from length-at-age data. Oceans (Roberts, 1986; Fig. 1). It ficult to count in both species, scale Juvenile hapuku are pelagic and supports significant fisheries off the most switch to a demersal habitat at annuli are unreliable for older fish, around 50 cm total length and at an Juan Fernández Islands, Chile, and and the ageing techniques have not estimated age of 3–4 years.