Gene-Regulatory Network Analysis of Ankylosing Spondylitis with a Single
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Detection of Interacting Transcription Factors in Human Tissues Using
Myšičková and Vingron BMC Genomics 2012, 13(Suppl 1):S2 http://www.biomedcentral.com/1471-2164/13/S1/S2 PROCEEDINGS Open Access Detection of interacting transcription factors in human tissues using predicted DNA binding affinity Alena Myšičková*, Martin Vingron From The Tenth Asia Pacific Bioinformatics Conference (APBC 2012) Melbourne, Australia. 17-19 January 2012 Abstract Background: Tissue-specific gene expression is generally regulated by combinatorial interactions among transcription factors (TFs) which bind to the DNA. Despite this known fact, previous discoveries of the mechanism that controls gene expression usually consider only a single TF. Results: We provide a prediction of interacting TFs in 22 human tissues based on their DNA-binding affinity in promoter regions. We analyze all possible pairs of 130 vertebrate TFs from the JASPAR database. First, all human promoter regions are scanned for single TF-DNA binding affinities with TRAP and for each TF a ranked list of all promoters ordered by the binding affinity is created. We then study the similarity of the ranked lists and detect candidates for TF-TF interaction by applying a partial independence test for multiway contingency tables. Our candidates are validated by both known protein-protein interactions (PPIs) and known gene regulation mechanisms in the selected tissue. We find that the known PPIs are significantly enriched in the groups of our predicted TF-TF interactions (2 and 7 times more common than expected by chance). In addition, the predicted interacting TFs for studied tissues (liver, muscle, hematopoietic stem cell) are supported in literature to be active regulators or to be expressed in the corresponding tissue. -

Table S1. List of Proteins in the BAHD1 Interactome
Table S1. List of proteins in the BAHD1 interactome BAHD1 nuclear partners found in this work yeast two-hybrid screen Name Description Function Reference (a) Chromatin adapters HP1α (CBX5) chromobox homolog 5 (HP1 alpha) Binds histone H3 methylated on lysine 9 and chromatin-associated proteins (20-23) HP1β (CBX1) chromobox homolog 1 (HP1 beta) Binds histone H3 methylated on lysine 9 and chromatin-associated proteins HP1γ (CBX3) chromobox homolog 3 (HP1 gamma) Binds histone H3 methylated on lysine 9 and chromatin-associated proteins MBD1 methyl-CpG binding domain protein 1 Binds methylated CpG dinucleotide and chromatin-associated proteins (22, 24-26) Chromatin modification enzymes CHD1 chromodomain helicase DNA binding protein 1 ATP-dependent chromatin remodeling activity (27-28) HDAC5 histone deacetylase 5 Histone deacetylase activity (23,29,30) SETDB1 (ESET;KMT1E) SET domain, bifurcated 1 Histone-lysine N-methyltransferase activity (31-34) Transcription factors GTF3C2 general transcription factor IIIC, polypeptide 2, beta 110kDa Required for RNA polymerase III-mediated transcription HEYL (Hey3) hairy/enhancer-of-split related with YRPW motif-like DNA-binding transcription factor with basic helix-loop-helix domain (35) KLF10 (TIEG1) Kruppel-like factor 10 DNA-binding transcription factor with C2H2 zinc finger domain (36) NR2F1 (COUP-TFI) nuclear receptor subfamily 2, group F, member 1 DNA-binding transcription factor with C4 type zinc finger domain (ligand-regulated) (36) PEG3 paternally expressed 3 DNA-binding transcription factor with -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

Pig Antibodies
Pig Antibodies gene_name sku Entry_Name Protein_Names Organism Length Identity CDX‐2 ARP31476_P050 D0V4H7_PIG Caudal type homeobox 2 (Fragment) Sus scrofa (Pig) 147 100.00% CDX‐2 ARP31476_P050 A7MAE3_PIG Caudal type homeobox transcription factor 2 (Fragment) Sus scrofa (Pig) 75 100.00% Tnnt3 ARP51286_P050 Q75NH3_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 271 85.00% Tnnt3 ARP51286_P050 Q75NH2_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 266 85.00% Tnnt3 ARP51286_P050 Q75NH1_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 260 85.00% Tnnt3 ARP51286_P050 Q75NH0_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 250 85.00% Tnnt3 ARP51286_P050 Q75NG8_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 266 85.00% Tnnt3 ARP51286_P050 Q75NG7_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 260 85.00% Tnnt3 ARP51286_P050 Q75NG6_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 250 85.00% Tnnt3 ARP51286_P050 TNNT3_PIG Troponin T, fast skeletal muscle (TnTf) Sus scrofa (Pig) 271 85.00% ORF Names:PANDA_000462 EMBL EFB13877.1OrganismAiluropod High mobility group protein B2 (High mobility group protein a melanoleuca (Giant panda) ARP31939_P050 HMGB2_PIG 2) (HMG‐2) Sus scrofa (Pig) 210 100.00% Agpat5 ARP47429_P050 B8XTR3_PIG 1‐acylglycerol‐3‐phosphate O‐acyltransferase 5 Sus scrofa (Pig) 365 85.00% irf9 ARP31200_P050 Q29390_PIG Transcriptional regulator ISGF3 gamma subunit (Fragment) Sus scrofa (Pig) 57 100.00% irf9 ARP31200_P050 Q0GFA1_PIG Interferon regulatory factor 9 Sus scrofa (Pig) -

NRF2 and the Ambiguous Consequences of Its Activation During Initiation and the Subsequent Stages of Tumourigenesis
cancers Review NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis Holly Robertson 1,2, Albena T. Dinkova-Kostova 1 and John D. Hayes 1,* 1 Jacqui Wood Cancer Centre, Division of Cellular Medicine, Ninewells Hospital and Medical School, University of Dundee, Dundee DD1 9SY, Scotland, UK; [email protected] (H.R.); [email protected] (A.T.D.-K.) 2 Wellcome Trust Sanger Institute, Wellcome Genome Campus, Cambridge CB10 1SA, UK * Correspondence: [email protected]; Tel.: +44-(0)-1382-383182 Received: 30 October 2020; Accepted: 27 November 2020; Published: 2 December 2020 Simple Summary: Transcription factor NRF2 controls expression of antioxidant and detoxification genes. Normally, the activity of NRF2 is tightly controlled in the cell, and is continuously adjusted to ensure that cells are protected against endogenous chemicals and environmental agents that perturb the intracellular antioxidant/pro-oxidant balance (i.e., redox) that must be maintained for them to grow and survive in an appropriate manner. This tight control of NRF2 is achieved by a repressor protein called KEAP1 that perpetually targets NRF2 protein for degradation under normal conditions, but is unable to do so when challenged with oxidants or thiol-reactive chemicals. In the context of cancer, it is well known that drugs that stimulate short-term and reversible activation of NRF2 can provide protection for a limited period against exposure to chemicals that cause cancer. However, it is also becoming widely recognised that permanent hyper-activation of NRF2 resulting from somatic mutations in the gene that encodes NRF2, or in genes associated with its degradation, is frequently observed in certain cancers and associated with poor outcome. -

Graph Complexity Analysis Identifies an ETV5 Tumor-Specific Network in Human and Murine Low-Grade Glioma." Plos One.13,5
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 Graph complexity analysis identifies na ETV5 tumor-specific network in human and murine low- grade glioma Yuan Pan Washington University School of Medicine in St. Louis Christina Duron Claremont Graduate University Erin C. Bush Claremont Graduate University Yu Ma Washington University School of Medicine in St. Louis Peter A. Sims Columbia University See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Pan, Yuan; Duron, Christina; Bush, Erin C.; Ma, Yu; Sims, Peter A.; Gutmann, David H.; Radunskaya, Ami; and Hardin, Johanna, ,"Graph complexity analysis identifies an ETV5 tumor-specific network in human and murine low-grade glioma." PLoS One.13,5. e0190001. (2018). https://digitalcommons.wustl.edu/open_access_pubs/6939 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Yuan Pan, Christina Duron, Erin C. Bush, Yu Ma, Peter A. Sims, David H. Gutmann, Ami Radunskaya, and Johanna Hardin This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/6939 RESEARCH ARTICLE Graph complexity analysis identifies an ETV5 tumor-specific network in human and murine low-grade glioma Yuan -

Pioneer Activity of an Oncogenic Fusion Transcription Factor at Inaccessible Chromatin
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.11.420232; this version posted June 7, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Title Pioneer activity of an oncogenic fusion transcription factor at inaccessible chromatin Authors Benjamin Sunkel1,6, Meng Wang1,6, Stephanie LaHaye2,6, Benjamin J. Kelly2, James R. Fitch2, FreDeric G. Barr3, Peter White2,4, Benjamin Z. Stanton1,4,5,7,* Author Affiliations 1 Nationwide Children’s Hospital, Center for Childhood Cancer and Blood Diseases, Columbus, OH 43205, USA 2 The Steve and Cindy Rasmussen Institute for Genomic Medicine, Nationwide Children’s Hospital, Columbus, OH 43205, USA 3 Laboratory of Pathology, National Cancer Institute, Bethesda, MD 20892, USA 4 Department of Pediatrics, The Ohio State University College of Medicine, Columbus, OH 43210, USA 5 Department of Biological Chemistry and Pharmacology, The Ohio State University College of Medicine, Columbus, OH 43210, USA 6These authors contributed eQually 7Lead contact *Correspondence: [email protected] (B.Z.S.) Summary Recent characterizations of pioneer transcription factors have leD to new insights into their structures anD patterns of chromatin recognition that are instructive for understanding their role in cell fate commitment and transformation. Intersecting with these basic science concepts, the iDentification of pioneer factors (PFs) fuseD together as Driver translocations in chilDhooD cancers raises questions of whether these fusions retain the funDamental ability to invade repressed chromatin, consistent with their monomeric PF constituents. -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function. -

Organ of Corti Size Is Governed by Yap/Tead-Mediated Progenitor Self-Renewal
Organ of Corti size is governed by Yap/Tead-mediated progenitor self-renewal Ksenia Gnedevaa,b,1, Xizi Wanga,b, Melissa M. McGovernc, Matthew Bartond,2, Litao Taoa,b, Talon Treceka,b, Tanner O. Monroee,f, Juan Llamasa,b, Welly Makmuraa,b, James F. Martinf,g,h, Andrew K. Grovesc,g,i, Mark Warchold, and Neil Segila,b,1 aDepartment of Stem Cell Biology and Regenerative Medicine, Keck Medicine of University of Southern California, Los Angeles, CA 90033; bCaruso Department of Otolaryngology–Head and Neck Surgery, Keck Medicine of University of Southern California, Los Angeles, CA 90033; cDepartment of Neuroscience, Baylor College of Medicine, Houston, TX 77030; dDepartment of Otolaryngology, Washington University in St. Louis, St. Louis, MO 63130; eAdvanced Center for Translational and Genetic Medicine, Lurie Children’s Hospital of Chicago, Chicago, IL 60611; fDepartment of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX 77030; gProgram in Developmental Biology, Baylor College of Medicine, Houston, TX 77030; hCardiomyocyte Renewal Laboratory, Texas Heart Institute, Houston, TX 77030 and iDepartment of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030; Edited by Marianne E. Bronner, California Institute of Technology, Pasadena, CA, and approved April 21, 2020 (received for review January 6, 2020) Precise control of organ growth and patterning is executed However, what initiates this increase in Cdkn1b expression re- through a balanced regulation of progenitor self-renewal and dif- mains unclear. In addition, conditional ablation of Cdkn1b in the ferentiation. In the auditory sensory epithelium—the organ of inner ear is not sufficient to completely relieve the block on Corti—progenitor cells exit the cell cycle in a coordinated wave supporting cell proliferation (9, 10), suggesting the existence of between E12.5 and E14.5 before the initiation of sensory receptor additional repressive mechanisms. -

Genetic Analyses of Human Fetal Retinal Pigment Epithelium Gene Expression Suggest Ocular Disease Mechanisms
ARTICLE https://doi.org/10.1038/s42003-019-0430-6 OPEN Genetic analyses of human fetal retinal pigment epithelium gene expression suggest ocular disease mechanisms Boxiang Liu 1,6, Melissa A. Calton2,6, Nathan S. Abell2, Gillie Benchorin2, Michael J. Gloudemans 3, 1234567890():,; Ming Chen2, Jane Hu4, Xin Li 5, Brunilda Balliu5, Dean Bok4, Stephen B. Montgomery 2,5 & Douglas Vollrath2 The retinal pigment epithelium (RPE) serves vital roles in ocular development and retinal homeostasis but has limited representation in large-scale functional genomics datasets. Understanding how common human genetic variants affect RPE gene expression could elu- cidate the sources of phenotypic variability in selected monogenic ocular diseases and pin- point causal genes at genome-wide association study (GWAS) loci. We interrogated the genetics of gene expression of cultured human fetal RPE (fRPE) cells under two metabolic conditions and discovered hundreds of shared or condition-specific expression or splice quantitative trait loci (e/sQTLs). Co-localizations of fRPE e/sQTLs with age-related macular degeneration (AMD) and myopia GWAS data suggest new candidate genes, and mechan- isms by which a common RDH5 allele contributes to both increased AMD risk and decreased myopia risk. Our study highlights the unique transcriptomic characteristics of fRPE and provides a resource to connect e/sQTLs in a critical ocular cell type to monogenic and complex eye disorders. 1 Department of Biology, Stanford University, Stanford, CA 94305, USA. 2 Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305, USA. 3 Program in Biomedical Informatics, Stanford University School of Medicine, Stanford 94305 CA, USA. 4 Department of Ophthalmology, Jules Stein Eye Institute, UCLA, Los Angeles 90095 CA, USA. -

International Journal of Phytopharmacology
183 Andrea Perra and Marta Anna Kowalik. / International Journal of Biological & Pharmaceutical Research. 2016; 7(4): 183-191. e- ISSN 0976 - 3651 Print ISSN 2229 - 7480 International Journal of Biological & Pharmaceutical Research Journal homepage: www.ijbpr.com IJBPR YES-ASSOCIATED PROTEIN (YAP) – A PROMISING THERAPEUTIC TARGET FOR HEPATOCELLULAR CARCINOMA Andrea Perra* and Marta Anna Kowalik Oncology and Molecular Pathology Unit, Department of Biomedical Sciences, University of Cagliari, via Porcell 4, 09124 Cagliari, Italy. ABSTRACT Hepatocellular carcinoma (HCC), the third leading cause of cancer mortality, carries a dismal prognosis and represents a major health problem. A better understanding of the molecular pathways involved in HCC development may represent an important approach for the improvement of the therapeutic strategies for this cancer. The Hippo signalling pathway, a growth- suppressive mechanism that antagonizes the transcriptional co-activator Yes-associated protein (YAP), has been recently found altered in human HCC. Moreover, using the Resistant-Hepatocyte (R-H) rat model, recently published data established that the Hippo pathway deregulation occurs already at the early stages of HCC development, making this pathway an important therapeutic target. Many strategies have been proposed to modulate the activity of the Hippo pathway, and in particular that of the final effector YAP, most of which require complex gene manipulations, useful to understand the functions of the pathway but difficult to apply on patients. One of the few drugs able to inhibit in vivo the pro-carcinogenic effect of YAP activation is verteporfin, which disrupts the formation of the complex between YAP and the TEAD transcription factors, causing a significant reduction of the number and size of preneoplastic foci induced in rats liver by R-H protocol. -
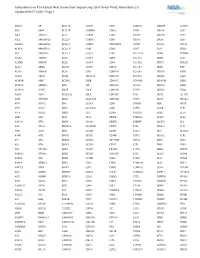
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1 ABCC3 AR BCL11A CANT1 CDK1 CMKLR1 DAB2IP DUSP9 ABI1 ARAF BCL11B CAPRIN1 CDK12 CNBP DACH1 E2F1 ABL1 ARFRP1 BCL2 CAPZB CDK2 CNOT2 DACH2 E2F3 ABL2 ARHGAP20 BCL2A1 CARD11 CDK4 CNTN1 DAXX EBF1 ABLIM1 ARHGAP26 BCL2L1 CARM1 CDK5RAP2 CNTRL DCLK2 ECT2L ACACA ARHGEF12 BCL2L11 CARS CDK6 COG5 DCN EDIL3 ACE ARHGEF7 BCL2L2 CASC5 CDK7 COL11A1 DDB1 EDNRB ACER1 ARID1A BCL3 CASP3 CDK8 COL1A1 DDB2 EED ACSBG1 ARID1B BCL6 CASP7 CDK9 COL1A2 DDIT3 EEFSEC ACSL3 ARID2 BCL7A CASP8 CDKL5 COL3A1 DDR2 EGF ACSL6 ARID5B BCL9 CAV1 CDKN1A COL6A3 DDX10 EGFR ACVR1 ARIH2 BCOR CBFA2T3 CDKN1B COL9A3 DDX20 EGR1 ACVR1B ARNT BCORL1 CBFB CDKN1C COMMD1 DDX39B EGR2 ACVR1C ARRDC4 BCR CBL CDKN2A COX6C DDX3X EGR3 ACVR2A ASMTL BDNF CBLB CDKN2B CPNE1 DDX41 EGR4 ADD3 ASPH BHLHE22 CBLC CDKN2C CPS1 DDX5 EIF1AX ADM ASPSCR1 BICC1 CCDC28A CDKN2D CPSF6 DDX6 EIF4A2 AFF1 ASTN2 BIN1 CCDC6 CDX1 CRADD DEK EIF4E AFF3 ASXL1 BIRC3 CCDC88C CDX2 CREB1 DGKB ELF3 AFF4 ASXL2 BIRC6 CCK CEBPA CREB3L1 DGKI ELF4 AGR3 ATF1 BLM CCL2 CEBPB CREB3L2 DGKZ ELK4 AHCYL1 ATF3 BMP4 CCNA2 CEBPD CREBBP DICER1 ELL AHI1 ATG13 BMPR1A CCNB1IP1 CEBPE CRKL DIRAS3 ELN AHR ATG5 BRAF CCNB3 CENPF CRLF2 DIS3 ELOVL2 AHRR ATIC BRCA1 CCND1 CENPU CRTC1 DIS3L2 ELP2 AIP ATL1 BRCA2 CCND2 CEP170B CRTC3 DKK1 EML1 AK2 ATM BRCC3 CCND3 CEP57 CSF1 DKK2 EML4 AK5 ATP1B4 BRD1 CCNE1 CEP85L CSF1R DKK4 ENPP2 AKAP12 ATP8A2 BRD3 CCNG1 CHCHD7 CSF3 DLEC1 EP300 AKAP6 ATR BRD4 CCT6B CHD2 CSF3R DLL1 EP400 AKAP9 ATRNL1 BRIP1 CD19 CHD4 CSNK1A1 DLL3