Gene Expression Microarray Analysis of the Sciatic Nerve of Mice with Diabetic Neuropathy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Par6c Is at the Mother Centriole and Controls Centrosomal Protein
860 Research Article Par6c is at the mother centriole and controls centrosomal protein composition through a Par6a-dependent pathway Vale´rian Dormoy, Kati Tormanen and Christine Su¨ tterlin* Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697-2300, USA *Author for correspondence ([email protected]) Accepted 3 December 2012 Journal of Cell Science 126, 860–870 ß 2013. Published by The Company of Biologists Ltd doi: 10.1242/jcs.121186 Summary The centrosome contains two centrioles that differ in age, protein composition and function. This non-membrane bound organelle is known to regulate microtubule organization in dividing cells and ciliogenesis in quiescent cells. These specific roles depend on protein appendages at the older, or mother, centriole. In this study, we identified the polarity protein partitioning defective 6 homolog gamma (Par6c) as a novel component of the mother centriole. This specific localization required the Par6c C-terminus, but was independent of intact microtubules, the dynein/dynactin complex and the components of the PAR polarity complex. Par6c depletion resulted in altered centrosomal protein composition, with the loss of a large number of proteins, including Par6a and p150Glued, from the centrosome. As a consequence, there were defects in ciliogenesis, microtubule organization and centrosome reorientation during migration. Par6c interacted with Par3 and aPKC, but these proteins were not required for the regulation of centrosomal protein composition. Par6c also associated with Par6a, which controls protein recruitment to the centrosome through p150Glued. Our study is the first to identify Par6c as a component of the mother centriole and to report a role of a mother centriole protein in the regulation of centrosomal protein composition. -

Centriole Overduplication Is the Predominant Mechanism Leading to Centrosome Amplification in Melanoma
Published OnlineFirst January 12, 2018; DOI: 10.1158/1541-7786.MCR-17-0197 Oncogenes and Tumor Suppressors Molecular Cancer Research Centriole Overduplication is the Predominant Mechanism Leading to Centrosome Amplification in Melanoma Ryan A. Denu1,2, Maria Shabbir3, Minakshi Nihal3, Chandra K. Singh3, B. Jack Longley3,4,5, Mark E. Burkard2,4, and Nihal Ahmad3,4,5 Abstract Centrosome amplification (CA) is common in cancer and can evaluated. PLK4 is significantly overexpressed in melanoma com- arise by centriole overduplication or by cell doubling events, pared with benign nevi and in a panel of human melanoma cell including the failure of cell division and cell–cell fusion. To assess lines (A375, Hs294T, G361, WM35, WM115, 451Lu, and SK-MEL- the relative contributions of these two mechanisms, the number of 28) compared with normal human melanocytes. Interestingly, centrosomes with mature/mother centrioles was examined by although PLK4 expression did not correlate with CA in most cases, immunofluorescence in a tissue microarray of human melanomas treatment of melanoma cells with a selective small-molecule PLK4 and benign nevi (n ¼ 79 and 17, respectively). The centrosomal inhibitor (centrinone B) significantly decreased cell proliferation. protein 170 (CEP170) was used to identify centrosomes with The antiproliferative effects of centrinone B were also accompa- mature centrioles; this is expected to be present in most centro- nied by induction of apoptosis. somes with cell doubling, but on fewer centrosomes with over- duplication. Using this method, it was determined that the major- Implications: This study demonstrates that centriole overdupli- ity of CA in melanoma can be attributed to centriole overduplica- cation is the predominant mechanism leading to centrosome tion rather than cell doubling events. -

Characterization of NIP2/Centrobin, a Novel Substrate of Nek2, and Its Potential Role in Microtubule Stabilization
2106 Research Article Characterization of NIP2/centrobin, a novel substrate of Nek2, and its potential role in microtubule stabilization Yeontae Jeong, Jungmin Lee, Kyeongmi Kim, Jae Cheal Yoo and Kunsoo Rhee* Department of Biological Sciences and Research Center for Functional Cellulomics, Seoul National University, Seoul 151-742, Korea *Author for correspondence (e-mail: [email protected]) Accepted 11 April 2007 Journal of Cell Science 120, 2106-2116 Published by The Company of Biologists 2007 doi:10.1242/jcs.03458 Summary Nek2 is a mitotic kinase whose activity varies during the associated with microtubules. Knockdown of NIP2 showed cell cycle. It is well known that Nek2 is involved in significant reduction of microtubule organizing activity, centrosome splitting, and a number of studies have cell shrinkage, defects in spindle assembly and abnormal indicated that Nek2 is crucial for maintaining the integrity nuclear morphology. Based on our results, we propose that of centrosomal structure and microtubule nucleation NIP2 has a role in stabilizing the microtubule structure. activity. In the present study, we report that NIP2, Phosphorylation may be crucial for mobilization of the previously identified as centrobin, is a novel substrate of protein to a new microtubule and stabilizing it. Nek2. NIP2 was daughter-centriole-specific, but was also found in association with a stable microtubule network of cytoplasm. Ectopic NIP2 formed aggregates but was Key words: NIP2, Centrobin, Nek2, Centrosome, Mitosis, dissolved by Nek2 into small pieces and eventually Microtubule Introduction through the phosphorylation-induced displacement of C-Nap1 Phosphorylation is one of major control mechanisms used from the centriolar ends (reviewed in Fry, 2002). -

S41467-019-10037-Y.Pdf
ARTICLE https://doi.org/10.1038/s41467-019-10037-y OPEN Feedback inhibition of cAMP effector signaling by a chaperone-assisted ubiquitin system Laura Rinaldi1, Rossella Delle Donne1, Bruno Catalanotti 2, Omar Torres-Quesada3, Florian Enzler3, Federica Moraca 4, Robert Nisticò5, Francesco Chiuso1, Sonia Piccinin5, Verena Bachmann3, Herbert H Lindner6, Corrado Garbi1, Antonella Scorziello7, Nicola Antonino Russo8, Matthis Synofzik9, Ulrich Stelzl 10, Lucio Annunziato11, Eduard Stefan 3 & Antonio Feliciello1 1234567890():,; Activation of G-protein coupled receptors elevates cAMP levels promoting dissociation of protein kinase A (PKA) holoenzymes and release of catalytic subunits (PKAc). This results in PKAc-mediated phosphorylation of compartmentalized substrates that control central aspects of cell physiology. The mechanism of PKAc activation and signaling have been largely characterized. However, the modes of PKAc inactivation by regulated proteolysis were unknown. Here, we identify a regulatory mechanism that precisely tunes PKAc stability and downstream signaling. Following agonist stimulation, the recruitment of the chaperone- bound E3 ligase CHIP promotes ubiquitylation and proteolysis of PKAc, thus attenuating cAMP signaling. Genetic inactivation of CHIP or pharmacological inhibition of HSP70 enhances PKAc signaling and sustains hippocampal long-term potentiation. Interestingly, primary fibroblasts from autosomal recessive spinocerebellar ataxia 16 (SCAR16) patients carrying germline inactivating mutations of CHIP show a dramatic dysregulation of PKA signaling. This suggests the existence of a negative feedback mechanism for restricting hormonally controlled PKA activities. 1 Department of Molecular Medicine and Medical Biotechnologies, University Federico II, 80131 Naples, Italy. 2 Department of Pharmacy, University Federico II, 80131 Naples, Italy. 3 Institute of Biochemistry and Center for Molecular Biosciences, University of Innsbruck, A-6020 Innsbruck, Austria. -
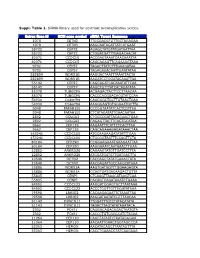
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

WO 2016/081450 Al 26 May 2016 (26.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/081450 Al 26 May 2016 (26.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every G01N33/574 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 15/06 1073 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 17 November 2015 (17.1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 62/08 1,445 18 November 2014 (18. 11.2014) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: BLUEPRINT MEDICINES CORPORA¬ GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TION [US/US]; 215 First Street, Cambridge, MA 02142 TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (US). -

Less Understood Issues: P21cip1 in Mitosis and Its Therapeutic Potential
Oncogene (2015) 34, 1758–1767 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc REVIEW Less understood issues: p21Cip1 in mitosis and its therapeutic potential N-N Kreis, F Louwen and J Yuan p21Cip1 is a multifunctional protein and a key player in regulating different cellular processes. The transcription of p21 is regulated by p53-dependent and -independent pathways. The expression of p21 is increased in response to various cellular stresses to arrest the cell cycle and ensure genomic stability. p21 has been shown to be a tumor suppressor and an oncogene as well. The function of p21 in mitosis has been proposed but not systematically studied. We have recently shown that p21 binds to and inhibits the activity of Cdk1/ cyclin B1, and is important for a fine-tuned mitotic progression. Loss of p21 prolongs the duration of mitosis and results in severe mitotic defects like chromosome segregation and cytokinesis failures promoting consequently genomic instability. Moreover, p21 is dramatically stabilized in mitotic tumor cells upon treatment with mitotic agents like paclitaxel or mitotic kinase inhibitors. Increased p21 is mainly localized in the cytoplasm and associates with cell survival indicating a crucial role of p21 in susceptibility to mitotic agents in tumor cells. In this review we will briefly summarize the structure and general physiological functions as well as regulation of p21, discuss in detail its role in mitosis and its potential to serve as a therapeutic target. Oncogene (2015) 34, 1758–1767; doi:10.1038/onc.2014.133; published online 26 May 2014 INTRODUCTION arguing about the role of p21 as a classical tumor suppressor. -

The Translational Landscape of the Splicing Factor SRSF1 and Its Role In
RESEARCH ARTICLE elifesciences.org The translational landscape of the splicing factor SRSF1 and its role in mitosis Magdalena M Maslon1†, Sara R Heras1,2†, Nicolas Bellora3‡, Eduardo Eyras3,4, Javier F Cáceres1* 1MRC Human Genetics Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, United Kingdom; 2GENYO, Centre for Genomics and Oncological Research, Pfizer/University of Granada/Andalusian Regional Government, Granada, Spain; 3Computational Genomics Group, Universitat Pompeu Fabra, Barcelona, Spain; 4Catalan Institution for Research and Advanced Studies (ICREA), Barcelona, Spain Abstract The shuttling serine/arginine rich (SR) protein SRSF1 (previously known as SF2/ASF) is a splicing regulator that also activates translation in the cytoplasm. In order to dissect the gene network that is translationally regulated by SRSF1, we performed a high-throughput deep sequencing analysis of polysomal fractions in cells overexpressing SRSF1. We identified approximately 1500 mRNAs that are translational targets of SRSF1. These include mRNAs encoding *For correspondence: Javier. proteins involved in cell cycle regulation, such as spindle, kinetochore, and M phase proteins, which [email protected] are essential for accurate chromosome segregation. Indeed, we show that translational activity of †These authors contributed SRSF1 is required for normal mitotic progression. Furthermore, we found that mRNAs that display equally to this work alternative splicing changes upon SRSF1 overexpression are also its translational targets, strongly suggesting that SRSF1 couples pre-mRNA splicing and translation. These data provide insights on Present address: ‡Laboratorio de Microbiología Aplicada y the complex role of SRSF1 in the control of gene expression at multiple levels and its implications Biotecnología, Centro Regional in cancer. -

The Greatwall Kinase Safeguards the Genome Integrity by Affecting the Kinome Activity in Mitosis
Oncogene (2020) 39:6816–6840 https://doi.org/10.1038/s41388-020-01470-1 ARTICLE The Greatwall kinase safeguards the genome integrity by affecting the kinome activity in mitosis 1,9 1 2 1,10 1,3 1 Xavier Bisteau ● Joann Lee ● Vinayaka Srinivas ● Joanna H. S. Lee ● Joanna Niska-Blakie ● Gifford Tan ● 1 1 4 5 1,6 7 Shannon Y. X. Yap ● Kevin W. Hom ● Cheng Kit Wong ● Jeongjun Chae ● Loo Chien Wang ● Jinho Kim ● 4 1,6 1,3,11 1,8 Giulia Rancati ● Radoslaw M. Sobota ● Chris S. H. Tan ● Philipp Kaldis Received: 17 June 2020 / Revised: 21 August 2020 / Accepted: 10 September 2020 / Published online: 25 September 2020 © The Author(s) 2020. This article is published with open access Abstract Progression through mitosis is balanced by the timely regulation of phosphorylation and dephosphorylation events ensuring the correct segregation of chromosomes before cytokinesis. This balance is regulated by the opposing actions of CDK1 and PP2A, as well as the Greatwall kinase/MASTL. MASTL is commonly overexpressed in cancer, which makes it a potential therapeutic anticancer target. Loss of Mastl induces multiple chromosomal errors that lead to the accumulation of micronuclei and multilobulated cells in mitosis. Our analyses revealed that loss of Mastl leads to chromosome breaks and abnormalities 1234567890();,: 1234567890();,: impairing correct segregation. Phospho-proteomic data for Mastl knockout cells revealed alterations in proteins implicated in multiple processes during mitosis including double-strand DNA damage repair. In silico prediction of the kinases with affected activity unveiled NEK2 to be regulated in the absence of Mastl. We uncovered that, RAD51AP1, involved in regulation of homologous recombination, is phosphorylated by NEK2 and CDK1 but also efficiently dephosphorylated by PP2A/B55. -

Identifying Human Interactors of SARS-Cov-2 Proteins and Drug Targets for COVID-19 Using Network-Based Label Propagation Arxiv:2
Identifying Human Interactors of SARS-CoV-2 Proteins and Drug Targets for COVID-19 using Network-Based Label Propagation Jeffrey N. Law1, Kyle Akers1, Nure Tasnina2, Catherine M. Della Santina3, Meghana Kshirsagar4, Judith Klein-Seetharaman5, Mark Crovella6, Padmavathy Rajagopalan7, Simon Kasif3, and T. M. Murali2,* 1Interdisciplinary Ph.D. Program in Genetics, Bioinformatics, and Computational Biology, Blacksburg, VA, USA 2Department of Computer Science, Virginia Tech, Blacksburg, VA, USA 3Department of Biomedical Engineering, Boston University, Boston, MA, USA 4AI for Good Lab, Microsoft, Redmond, WA, USA 5Department of Chemistry, Colorado School of Mines, Golden, CO USA 6Department of Computer Science, Boston University, Boston, MA, USA 7Department of Chemical Engineering, Virginia Tech, Blacksburg, VA, USA *Corresponding author. Email: [email protected] Motivated by the critical need to identify new treatments for COVID- arXiv:2006.01968v2 [q-bio.MN] 22 Jun 2020 19, we present a genome-scale, systems-level computational approach to prioritize drug targets based on their potential to regulate host- virus interactions or their downstream signaling targets. We adapt and specialize network label propagation methods to this end. We demonstrate that these techniques can predict human-SARS-CoV- 2 protein interactors with high accuracy. The top-ranked proteins 1 that we identify are enriched in host biological processes that are po- tentially coopted by the virus. We present cases where our method- ology generates promising insights such as the potential role of HSPA5 in viral entry. We highlight the connection between en- doplasmic reticulum stress, HSPA5, and anti-clotting agents. We identify tubulin proteins involved in ciliary assembly that are tar- geted by anti-mitotic drugs. -

Ncomms2517.Pdf
ARTICLE Received 17 May 2012 | Accepted 17 Jan 2013 | Published 26 Feb 2013 DOI: 10.1038/ncomms2517 LGALS3BP regulates centriole biogenesis and centrosome hypertrophy in cancer cells Marie-Laure Fogeron1,w,*, Hannah Mu¨ller1,*, Sophia Schade1, Felix Dreher1, Verena Lehmann1, Anne Ku¨hnel1, Anne-Kathrin Scholz1, Karl Kashofer2, Alexandra Zerck1, Beatrix Fauler1, Rudi Lurz1, Ralf Herwig1, Kurt Zatloukal2, Hans Lehrach1, Johan Gobom1,w, Eckhard Nordhoff1,w & Bodo M.H. Lange1,3 Centrosome morphology and number are frequently deregulated in cancer cells. Here, to identify factors that are functionally relevant for centrosome abnormalities in cancer cells, we established a protein-interaction network around 23 centrosomal and cell-cycle regulatory proteins, selecting the interacting proteins that are deregulated in cancer for further studies. One of these components, LGALS3BP, is a centriole- and basal body-associated protein with a dual role, triggering centrosome hypertrophy when overexpressed and causing accumulation of centriolar substructures when downregulated. The cancer cell line SK-BR-3 that overexpresses LGALS3BP exhibits hypertrophic centrosomes, whereas in seminoma tissues with low expression of LGALS3BP, supernumerary centriole-like structures are present. Centrosome hypertrophy is reversed by depleting LGALS3BP in cells endogenously over- expressing this protein, supporting a direct role in centrosome aberration. We propose that LGALS3BP suppresses assembly of centriolar substructures, and when depleted, causes accumulation of centriolar complexes comprising CPAP, acetylated tubulin and centrin. 1 Department of Vertebrate Genomics, Max-Planck Institute for Molecular Genetics, 14195 Berlin, Germany. 2 Institute of Pathology, Medical University of Graz, Graz 8036, Austria. 3 Alacris Theranostics GmbH, Fabeckstr. 60–62, 14195, Berlin, Germany. * These authors contributed equally to this work.